















Background
Dilated cardiomyopathy (DCM) is currently defined by the presence of left ventricular (LV) or biventricular dilatation and systolic dysfunction in the absence of abnormal loading conditions (hypertension, valve disease) or coronary artery disease sufficient to cause global systolic impairment.
At the time of diagnosis of DCM, acquired causes should be investigate, as well as a search for red flags such as early rhythm disorders or a positive family history of cardiomyopathy, suggesting the presence of a genetic aetiology that may lead to an appropriated diagnosis, risk stratification, treatment and familial surveillance.
Timeline
2010 – Presentation in a private clinic due to heart failure with reduced ejection fraction (HFrEF). He received a diagnosis of probably alcoholic cardiomyopathy. First-degree atrioventricular (AV) block.
Month 6 – 24h-Holter revealed the presence of premature ventricular complexes (PVC) and non-sustained ventricular tachycardia (NSVT).
2012 – Admission due to complete heart block. Heart failure with reduced ejection fraction (LVEF) 45%. He received a cardiac resynchonization therapy-pacemacker (CRT-P). Atrial fibrillation (AF) onset.
2015 – Heart failure admission. CRT-P non-responder due to high PVC burden. He was referred to the inherited cardiovascular conditions clinic. Next-generation sequencing (NGS) ordered.
Month 1 - The patient experienced sudden cardiac death.
Month 2 - Genetic sequencing revealed a frameshit mutation in LMNA (p.Trp514*).
Case Presentation
A 40-year-old male with a personal history of long-term heavy alcohol use, whose father died aged 48 due to idiopathic DCM during cardiac transplantation surgery, presented to a private clinic in 2010 due to exertional dyspnea. The patient was diagnosed with HFrEF and DCM probably related with alcohol abuse. At that moment, his electrocardiogram (ECG) showed sinus rhythm at 58 bpm, first-degree atrioventricular block with 480 ms and narrow QRS with low voltage in limb leads. Blood test showed normal (61 mg/dL) serum creatine kinase (CK) and mild elevated brain natriuretic peptide (370 mg/dL). The patient stopped drinking alcohol and started with Ramipril 2.5 mg oad, Bisoprolol 2.5 mg oad and Eplerenone 25 mg oad.
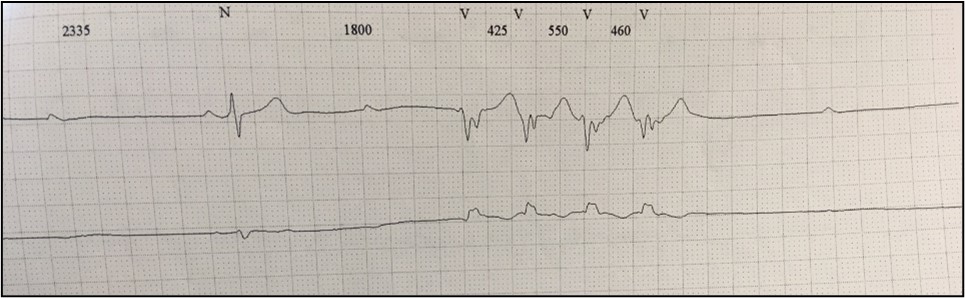
The following year, he was free of dyspnea (NYHA class I/IV), with an improvement of LVEF (45%). A 24h-Holter showed low density PVCs, two episodes of NSVT and a supraventricular extrasystoles burden of 10%, as well as episodes of nocturnal Wenckebach AV block (Figure 1).
In 2012 the patient was admitted presenting with dyspnea on moderate exertion. His ECG showed complete heart block with a narrow QRS rhythm at 40 bpm, which persisted after Bisoprolol washout. Also, a significant burden of PVC and NSVT episodes were detected during the monitoring (Figure 2).

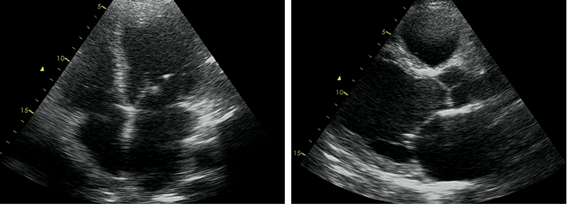
The transthoracic echocardiography showed a slightly dilated (115/70 mL) left ventricle (LV), with normal wall thickness, global hypokinesia and mildly reduced LVEF (45%). The left atrium was dilated (45 mm). Also, the right ventricle was dilated (45 mm) with preserved systolic function (Figure 3).
Due to the previous history of alcoholic cardiomyopathy, a mild impaired LVEF and a complete heart block, the patient underwent CRT-P implantation. Prior to discharge, the patient experienced the occurrence of AF, which few months later progressed to permanent AF.
During the following years he remained with mild symptoms (NYHA II/IV). CRT-P was ineffective due to frequent PVCs conditioning low biventricular pacing percentage. Frequent PVC were refractory to beta-blockers at maximum tolerated doses. Amiodarone was started but had to be discontinued because of patient preferences. This occasioned a heart failure admission during 2015.
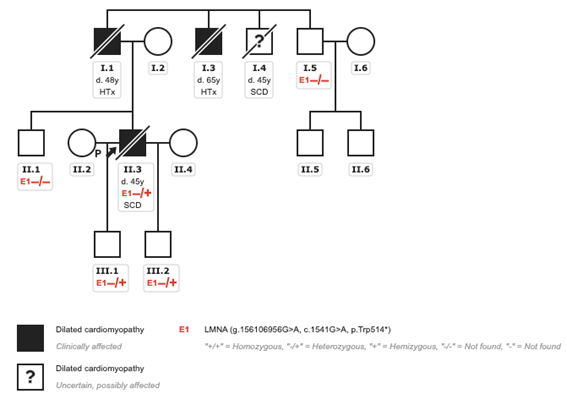
After this episode, he was referred for the first time to the inherited cardiovascular conditions clinic (ICC). A three generation pedigree was obtained (Figure 4), showing two cases of heart transplantation and a sudden cardiac death (SCD) in a paternal uncle. In our case, although DCM was initially attributed to alcohol consumption, the patient presented a positive family history of heart disease at young age and arrhythmic disturbances pointing to a LMNA genetic substrate. Therefore, a NGS panel for DCM was requested.
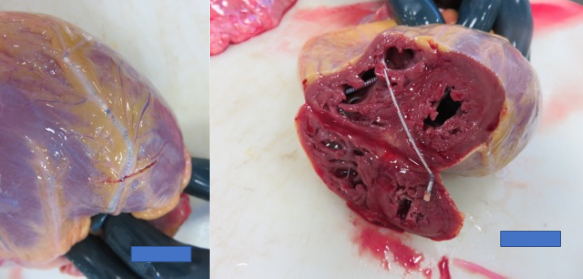
One month after the consultation and while awaiting the results of the genetic study, the patient suddenly died (Figure 5). Some weeks later, the genetic study reported a new pathogenic p.Trp514* frameshit mutation in the LMNA gene. Cascade screening identified this pathogenic mutation in his two sons.
Discussion
Here we have described the case of a young man with familial DCM due to a pathogenic LMNA mutation, initially attributed to alcohol abuse, who suffered a potentially preventable SCD. This clinical case emphasizes the importance of a guideline-directed diagnostic work-up and treatment in DCM.
DCM accounts for 30–40% of all heart failure cases in large clinical trials and is the leading cause of heart transplantation. It is genetically heterogeneous, and DCM genes encode proteins of broad cellular functions (cytoskeletal, sarcomeric, mitochondrial, desmosomal, nuclear membrane, and RNA-binding proteins). (1). Thus, the pathological mechanisms that lead to DCM are diverse. Among the 12 genes classified as having definitive evidence (BAG3, DES, FLNC, MYH7, PLN, RBM20, SCN5A, TNNC1, TNNT2, TTN). (2) One of the most frequent are LMNA missense and truncating mutations, accounting for 5% to 8% of genetic DCM. The single LMNA gene encodes lamins A and C, nuclear proteins implicated in many different cellular processes from regulating gene expression, mechanosensing, DNA replication, and nuclear to cytoplasmic transport. (1)
Clinically, LMNA-related DCM is an autosomal dominant severe heart disease characterized by the presence of LVEF impairment, progressive cardiac conduction disease (CCD) and/or supraventricular or ventricular arrhythmias such AF, that may precede the DCM phenotype. Occasionally, it may be associated with clinical myopathy, which is usually associated with elevated serum CK. This condition usually presents in early to mid-adulthood. (3)
The diagnosis is established in a proband with suggestive findings and a pathogenic variant in heterozygosity in LMNA. In suspected LMNA-related DCM, the initial clinical work-up should include a three to four generation family history (familial history of DCM, CCD, SCD, heart transplantation, AF or pacemaker implantation at early age, and neuromuscular disease), blood tests (creatin-kinase), ECG, 24h-Holter monitor and echocardiography. Cardiac magnetic resonance basal to midseptal midmyocardial late gadolinium enhancement appears to be a common and early finding correlating with CCD and ventricular arrhythmias (VA). In this case, the patient presented several red flags that characterised the LMNA-related DCM (family history of DCM, low voltage ECG, CCD and AF at young age, NSVT), and therefore earlier genetic testing was clearly recommended. (3,4)
Regarding the risk stratification, because it is associated with an increased risk of SCD, whether or not there is a LVEF £ 35%, and this may be the debut manifestation, several predictors of life-threatening VA have been evaluated. Thus, recently a risk calculator to predict the risk of life-threatening VA at 5 years has been developed. This calculator gives higher risk to male sex, non-missense LMNA mutation, high degree AV block, NSVT, LVEF < 45% at first evaluation. According to the 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of SCD, in patients with a 5-year estimated risk ≥10% with this calculator and a manifest cardiac phenotype (NSVT, LVEF < 50%, or AV conduction delay), a primary prevention implantable cardioverter-defibrillator (ICD) implantation should be considered. (5, 6) On the other hand, in a recently published Spanish cohort of 140 patients, they only found a LVEF < 45% and the presence of NSVT as predictors of major arrhythmic events (SCD or ICD shock). (7)
Based on the above information, the patient was a high-risk case of SCD that required CRT-D implantation and may have prevented sudden death.
Conclusions
LMNA-related DCM is a severe disease characterised by LVEF impairment, progressive CCD, supraventricular or ventricular arrhythmias and increased risk of SCD beyond the classical ejection fraction threshold for ICD implantation.
Careful diagnostic work-up, including early genetic testing in DCM, have to be considered in order to identify the underlying substrate, leading to a patient-tailored risk stratification and treatment, such as a more personalized approach to ICD-implantation.