















Keywords
Cardiac myosin inhibitors, gene therapy, hypertrophic cardiomyopathy, new drugs
Abbreviation list
HCM: hypertrophic cardiomyopathy
LVOTO: left ventricular outflow tract obstruction
Take-home messages
- Novel pharmacologic agents, particularly cardiac myosin inhibitors, represent a shift from symptom-focused therapy to disease-modifying treatment in HCM.
- The strongest evidence in favour of these new drugs is in obstructive HCM, where improvements in outflow obstruction, symptoms, and biomarker profiles have been demonstrated.
- Treatment options in nonobstructive HCM are still limited.
- Emerging strategies, such as gene therapy, hold promise for truly causative treatment.
Introduction
Hypertrophic cardiomyopathy (HCM) is recognised as the most common inherited cardiac disease. It is characterised by left ventricular hypertrophy that cannot be explained by other factors. Left ventricular outflow tract obstruction (LVOTO) is a common and clinically important feature of the disease [1].
The condition most frequently arises from pathogenic variants in genes encoding sarcomeric proteins. These genetic variants impair normal sarcomere structure and function, leading to molecular, histological, and morphological changes that disrupt myocardial performance [1,2]. Clinically, HCM is a progressive disorder associated with complications such as atrial fibrillation, heart failure, stroke, and sudden cardiac death [2].
Historically, treatment strategies have centred on symptom control using pharmacologic and invasive measures. However, advances in pharmacotherapy have begun to shift this landscape, with the introduction of agents that directly target sarcomeric dysfunction. This article reviews novel and emerging pharmacologic therapies for HCM.
Central Figure. Current, new, and emerging pharmacologic therapies in hypertrophic cardiomyopathy.
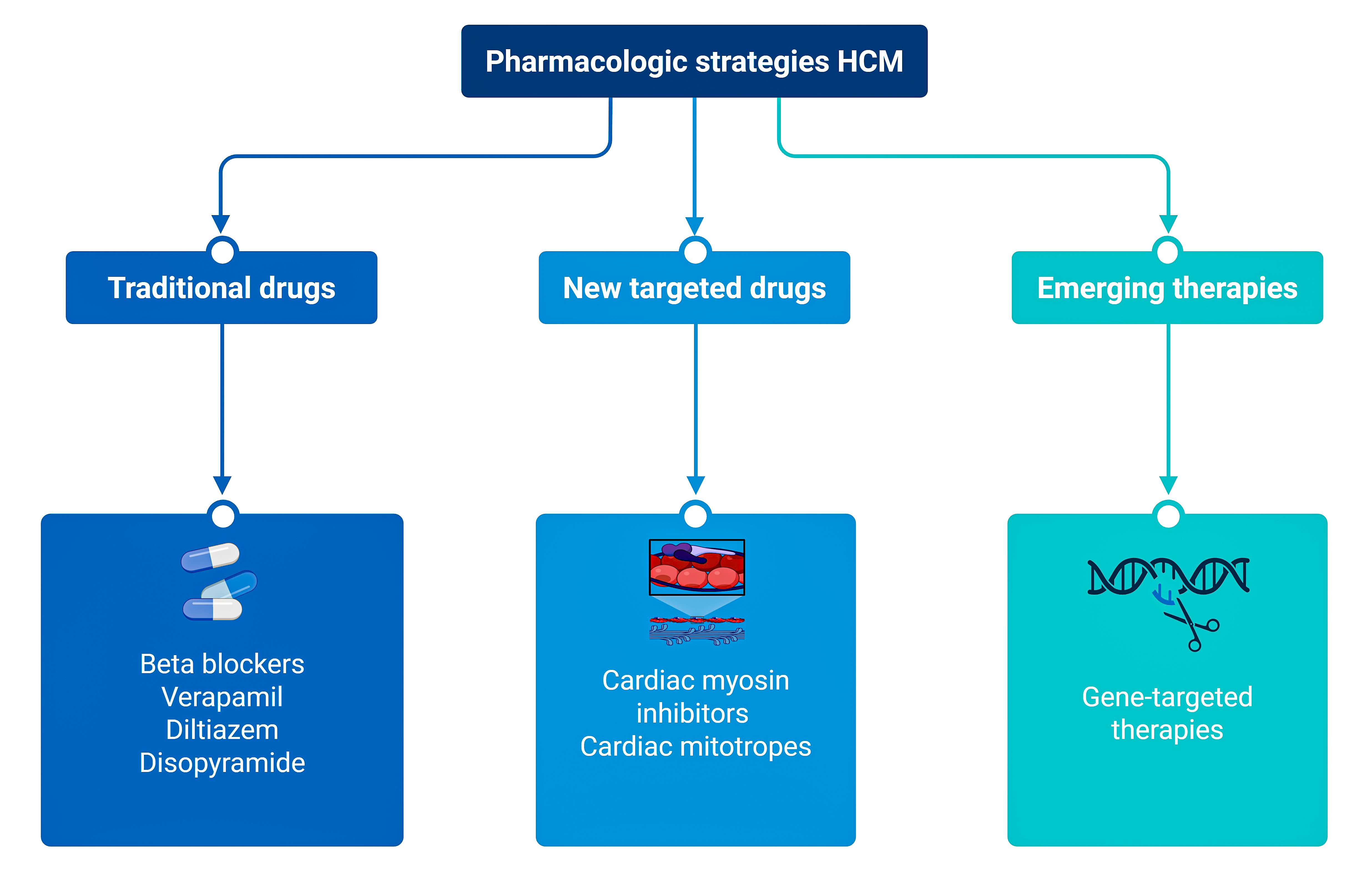
Overview of current and emerging pharmacologic therapies in hypertrophic cardiomyopathy (HCM). Traditional therapies primarily provide symptomatic relief by modulating heart rate, contractility, and left ventricular outflow tract obstruction. More recent mechanism-based therapies target sarcomere function and myocardial energetics. Emerging next-generation approaches aim to directly correct or prevent the molecular drivers of disease, potentially enabling true disease modification or prevention. Created with BioRender.com
Standard medical management of HCM
The cornerstone of pharmacologic therapy in HCM has traditionally focused on alleviating symptoms and preventing complications. For symptomatic patients with preserved ejection fraction, regardless of LVOTO, beta blockers and nondihydropyridine calcium channel blockers (verapamil, diltiazem) and disopyramide have been considered first-line treatments. The main aim of first-line therapy is to improve left ventricular filling and reduce LVOTO [1,2].
In patients with obstructive HCM, treatment options beyond first-line pharmacologic therapy have traditionally included invasive septal reduction procedures. These interventions have been reserved for symptomatic individuals who continue to exhibit significant obstruction despite optimal medical management. When symptoms persist in patients without LVOTO despite first-line therapy, oral diuretics may be introduced for further symptom management [2].
Although commonly employed in clinical practice, conventional therapies often fail to adequately relieve symptoms and may be poorly tolerated [2]. Additionally, current evidence does not demonstrate that these medications modify disease progression in HCM, improve exercise capacity, or decrease mortality [1,2].
Alongside standard medical treatment for control of symptoms, a critical aspect of HCM management involves risk stratification to guide prophylactic interventions for sudden cardiac death, including implantable cardioverter-defibrillator therapy, stroke prevention with anticoagulants in patients with atrial fibrillation, and the management of disease-related comorbidities [2].
Ongoing trials continue to refine the use of conventional pharmacologic therapies in HCM. The TEMPO II randomised crossover trial compares the effects of bisoprolol and verapamil on symptoms and functional outcomes in symptomatic nonobstructive HCM, aiming to clarify the optimal use of first-line therapy [3].
Pathophysiology and rationale for targeted therapy
In HCM, variants in genes encoding sarcomere proteins, most frequently MYH7 and MYBPC3, disrupt the normal sarcomere function. MYH7 variants alter the structure and function of the myosin molecule, increasing the proportion of myosin heads engaged with actin, while MYBPC3 mutations reduce the inhibitory modulation of myosin-actin interactions. Normally, myosin heads cycle between a relaxed, energy-saving state and an active, force-generating state. In HCM, more heads remain active, resulting in hypercontractility due to excessive force generation, impaired relaxation from incomplete detachment during diastole, and increased energy consumption because adenosine triphosphate (ATP) is continuously used by active cross-bridges [4]. Thin filament mutations can further increase the sensitivity of the contractile apparatus to calcium, driving energy use even higher and contributing to disturbances in normal energy pathways, including greater oxidative stress and reduced metabolic efficiency [4]. Notably, these molecular abnormalities can precede overt structural changes [4]. This molecular understanding provides the basis for novel drug therapies in HCM, including cardiac myosin inhibitors.
The consequences of disease progression may ultimately result in LVOTO, hypertrophy, cardiomyocyte disarray, interstitial fibrosis, and ventricular remodelling [1]. Accordingly, emerging therapeutic strategies aim to target hypercontractility and LVOTO, improve myocardial energetics, and modulate or reverse myocardial remodelling.
New pharmacologic therapies in HCM
The shortcomings of traditional symptomatic therapies have driven the development of disease-modifying agents, directly targeting the molecular mechanisms of HCM.
Sarcomere-targeted therapies
Cardiac myosin inhibitors represent a new class of drugs that reversibly inhibit cardiac myosin adenosine triphosphate. Mavacamten is the first drug in this class to reach routine clinical practice, having received regulatory approval in the United States in 2022 and in Europe in 2023. Aficamten, a next-generation inhibitor with greater selectivity for a distinct allosteric site on the myosin molecule, has completed Phase III studies in obstructive HCM, is now awaiting regulatory assessment, and is currently being evaluated in Phase III trials for nonobstructive disease. Delocamten (MYK-224), another compound in this class, is undergoing Phase II evaluation [5].
The key clinical trials of cardiac myosin inhibitors in HCM are summarised in Table 1. Mavacamten, the most extensively studied drug in this class, is an allosteric, selective, reversible inhibitor of cardiac myosin ATP phosphohydrolase (ATPase) [5]. Aficamten has a similar mechanism of action, but shows higher selectivity for the cardiac myosin active site and shows pharmacokinetic advantages, including a shorter half-life and improved tolerability [5,6].
Table 1. Key studies of cardiac myosin inhibitors in hypertrophic cardiomyopathy.
|
Study |
Population |
Drug |
Design |
N |
Duration |
NYHA |
Primary endpoint |
Outcome |
Key secondary endpoints |
|
EXPLORER-HCM [7] |
Obstructive |
Mavacamten |
Double-blind, randomised, placebo-controlled |
251 |
30 weeks |
II–III |
pVO₂ increase ≥1.5 mL/kg/min + ≥1 NYHA class improvement |
Positive |
↓ LVOT gradient, ↑ KCCQ-CSS, ↓ NT-proBNBP, ↓ hs-cTnI |
|
VALOR-HCM [8] |
Obstructive |
Mavacamten |
Double-blind, randomised, placebo-controlled |
112 |
56 weeks |
III–IV |
Reduction in SRT eligibility |
Positive |
↓ LVOT gradient, ↓ NYHA class |
|
MAVA-LTE [9] |
Obstructive |
Mavacamten |
Open-label extension |
231 |
252 weeks |
II–III |
Safety |
Favourable safety |
↓ LVOT gradient, ↓ NT-proBNP, ↓ NYHA class |
|
SEQUOIA-HCM [10] |
Obstructive |
Aficamten |
Double-blind, randomised vs placebo |
282 |
24 weeks |
II–III |
pVO₂ |
Positive |
↓ LVOT gradient, ↓ NYHA class, ↑ KCCQ-CSS |
|
FOREST-HCM [11] |
Obstructive |
Aficamten |
Open-label |
213 |
Ongoing (48-week analysis) |
II–III |
Safety |
Favourable safety |
↓ LVOT gradient, ↓ NT-proBNP, ↓ hs-cTnI |
|
MAPLE-HCM [12] |
Obstructive |
Aficamten |
Double-blind, randomised vs metoprolol |
175 |
24 weeks |
II–III |
pVO₂ |
Positive |
↓ LVOT gradient, ↓ NT-proBNP, ↑ KCCQ-CSS |
|
MAVERICK-HCM [13] |
Non- obstructive |
Mavacamten |
Double-blind, randomised, placebo-controlled |
59 |
16 weeks |
II–III |
Safety |
Favorable safety |
↓ NT-proBNP, ↓ hs-cTnI; pVO₂ and NYHA unchanged |
|
ODYSSEY-HCM [14,15] |
Non- obstructive |
Mavacamten |
Double-blind, randomised, placebo-controlled |
580 |
48 weeks |
II–III |
pVO₂, KCCQ-CSS |
Neutral (primary endpoints not met) |
↓ NT-proBNP, ↓HCMSQ shortness-of-breath score; VE/VCO₂ and NYHA class unchanged |
|
ACACIA-HCM [16] |
Non- obstructive |
Aficamten |
Double-blind, randomised, placebo-controlled |
420 |
36 weeks |
II–III |
KCCQ-CSS |
Ongoing |
pVO₂, VE/VCO₂, NYHA class, NT-proBNP, LAVI, MACE |
|
REDWOOD-HCM (Cohort 4) [6] |
Non- obstructive |
Aficamten |
Open-label, (phase 2) |
41 |
10 weeks |
II–III |
Safety + tolerability |
Favourable safety |
↑ NYHA, ↑ KCCQ-CSS, ↓ NT-proBNP, ↓ hs-cTnI, modest ↓ LVEF |
|
SCOUT-HCM [17] |
Obstructive(12 to <18 years) |
Mavacamten |
Double-blind, randomised, placebo-controlled |
40 |
28 weeks |
II–III |
Change in Valsalva LVOT gradient |
Ongoing |
Exercise/rest LVOT gradient, pVO₂, PK, safety |
|
CEDAR-HCM [18] |
Obstructive (children ≥6 years) |
Aficamten |
Double-blind, randomised, placebo-controlled |
≈65 |
12 weeks |
≥II |
Change in Valsalva LVOT gradient |
Ongoing |
Resting LVOT gradient, biomarkers, NYHA, PK, safety |
CPET: Cardiopulmonary Exercise Test; HCM: hypertrophic cardiomyopathy; hs-cTnI: high-sensitivity cardiac troponin I; KCCQ-CSS: Kansas City Cardiomyopathy Questionnaire–Clinical Summary Score; LAVI: left atrial volume index; LVEF: left ventricular ejection fraction; LVOT: left ventricular outflow tract; MACE: major adverse cardiovascular events; NT-proBNP: N-terminal pro-B-type natriuretic peptide; NYHA: New York Heart Association; PK: pharmacokinetics; pVO₂: peak oxygen consumption; SRT: septal reduction therapy; VE/VCO₂: ventilatory efficiency
Outcome definitions
Positive: primary endpoint met
Neutral: primary endpoint not met
Favourable safety: safety endpoint met without significant adverse cardiac effects
In patients with symptomatic obstructive HCM, mavacamten has demonstrated substantial clinical benefits, including marked reductions in LVOT gradients and corresponding improvements in exercise capacity, cardiac biomarkers, and symptoms, as shown in EXPLORER-HCM [7], MAVA-LTE [9], and EXPLORER-CN [19]. It has also been associated with a markedly reduced need for invasive septal reduction therapy. In VALOR-HCM, after 16 weeks of treatment, only 18% of patients receiving mavacamten met criteria for or chose to undergo septal reduction therapy, compared with 77% in the placebo group (P<0.0001) [8]. Additionally, FOREST-HCM substudies from EXPLORER-HCM have demonstrated reductions in left ventricular mass index and improvements in diastolic function following treatment with mavacamten [20,21].
In contrast, the benefits of cardiac myosin inhibitors in nonobstructive HCM are less well established. In the ODYSSEY-HCM trial, mavacamten failed to improve peak oxygen uptake or symptoms in this population and was associated with a higher incidence of reductions in left ventricular ejection fraction, despite significant reductions in NT-proBNP and cardiac troponin levels in MAVERICK-HCM [13,14,15].
Aficamten has shown similar clinical benefits in obstructive HCM, with significant reductions in LVOT gradients, improvements in exercise capacity, and alleviation of symptoms in the SEQUOIA-HCM trial [10]. Its efficacy in nonobstructive HCM is being evaluated in the ongoing ACACIA-HCM study [16,19]. The MAPLE-HCM trial, which directly compared aficamten with metoprolol in obstructive HCM, found superiority of aficamten monotherapy when evaluating peak oxygen uptake, LVOT gradients, and symptom burden [12].
In addition, paediatric studies are underway to evaluate aficamten and mavacamten in children and adolescents with obstructive HCM [17,18].
In terms of clinical implications, cardiac myosin inhibitors represent a noninvasive, disease-specific therapeutic option for obstructive HCM and may delay or lessen the need for septal reduction procedures, particularly in patients who continue to have LVOTO despite optimal conventional therapy. Current guidelines, including the ACC/AHA and ESC recommendations, now incorporate mavacamten for symptomatic obstructive HCM that is insufficiently controlled with first-line treatment. The principal safety concern with these agents is excessive reduction in left ventricular systolic function, and the treatment necessitates regular echocardiographic monitoring. A proportion of patients experience LVEF reductions that require dose adjustment or temporary interruption, and persistent systolic dysfunction mandates discontinuation [2]. Interindividual variability in drug metabolism, particularly related to CYP2C19 genotypes, affects dosing and safety, and genotyping is recommended to identify poor metabolisers [5].
Despite encouraging progress, several uncertainties remain for the future use of cardiac myosin inhibitors, particularly the durability of effect and long-term outcomes, applicability to nonobstructive HCM, cost-effectiveness, and optimal patient selection. Moreover, randomised controlled trials directly comparing cardiac myosin inhibitors with septal reduction therapy are lacking.
Beyond cardiac myosin inhibitors, additional sarcomere-targeted therapies are in development. EDG-7500 is a selective cardiac sarcomere modulator, designed to modulate the cardiac sarcomere to slow early contraction velocity and improve cardiac relaxation and filling. The drug is currently being studied in an open-label phase II trial involving patients with either obstructive or nonobstructive HCM [22].
Cardiac mitotropes
Recognition of the impaired myocardial energetics in HCM has spurred interest in therapies that shift substrate use, enhance efficiency, or reduce myocardial oxygen consumption. Ninerafaxstat is a cardiac mitotrope developed to restore myocardial energy balance [23]. Its effect is achieved by inhibiting the final enzyme (3-ketoacyl-CoA thiolase) involved in mitochondrial breakdown of long-chain fatty acids. This diversion away from fatty acid use promotes greater reliance on glucose oxidation, which generates ATP more efficiently and requires less oxygen, particularly when oxygen delivery is limited, thereby increasing myocardial metabolic efficiency [23].
The IMPROVE-HCM phase 2 trial assessed the safety and efficacy of ninerafaxstat in symptomatic patients with nonobstructive HCM. Although most prespecified efficacy endpoints were not met, ninerafaxstat showed signs of clinical benefit, including improved ventilatory efficiency and improvements in quality of life scores in patients with lower baseline scores. Treatment was also associated with favourable trends in natriuretic peptides and left atrial size without affecting troponin levels or LVEF. Importantly, the drug showed good tolerability and no reductions in LVEF [23]. On the basis of this safety profile and the metabolic and symptomatic signals detected, a phase 2B study (FORTITUDE-HCM) is underway to assess the clinical efficacy of ninerafaxstat in symptomatic nonobstructive HCM [24].
Non-sarcomere-targeted adjunctive therapies
Beyond sarcomere dysfunction, neurohormonal activation and myocardial remodelling are increasingly recognised as contributors to disease progression in HCM. This has led to interest in renin–angiotensin–aldosterone system inhibition as a potential strategy to attenuate hypertrophy and fibrosis. The safety and efficacy of the angiotensin II receptor blocker valsartan were evaluated in a multicentre, randomised, double-blind, placebo-controlled phase II trial enrolling 178 patients with early-stage sarcomeric HCM. Participants were randomised to receive valsartan or placebo for two years. The primary endpoint was a composite z-score integrating standardised changes in left ventricular wall thickness, mass, and volume, left atrial volume, tissue Doppler indices, and high-sensitivity troponin T and N-terminal pro-B-type natriuretic protein. Treatment with valsartan resulted in a significant improvement in the composite score compared with placebo, indicating favourable effects on cardiac structure and function, and was well-tolerated [25]. These findings suggest that an angiotensin II receptor blockade may attenuate disease evolution when initiated early in the course of sarcomeric HCM. In contrast, an earlier randomised trial of losartan showed no significant effect on left ventricular mass or disease progression in patients with overt HCM, even when applying a composite z-score endpoint as used in the VANISH trial [26,27]. On this basis, the 2024 ACC/AHA guidelines have expanded treatment considerations to include valsartan in younger patients (≤45 years) with pathogenic sarcomeric variants and a mild phenotype, to slow disease progression (class 2 recommendation) [2].
Myocardial fibrosis is a hallmark feature of HCM and contributes to diastolic dysfunction, ventricular arrhythmias, and adverse clinical outcomes [1]. In a randomised, double-blind, placebo-controlled trial, 61 patients with nonobstructive HCM were assigned to receive the mineralocorticoid receptor antagonist eplerenone or placebo for 12 months. The primary endpoint was native T1 time on cardiac magnetic resonance imaging, used as a marker of diffuse myocardial fibrosis. Treatment with eplerenone was associated with a significant reduction in native T1 time from baseline compared to placebo. Although the between-group difference did not reach statistical significance, the direction of effect was consistent with a reduction in diffuse myocardial fibrosis. No significant improvements were observed in exercise capacity or echocardiographic indices of diastolic function [28]. Overall, these data suggest that eplerenone may influence myocardial fibrosis in HCM. However, the modest sample size and lack of demonstrable functional benefit highlight the need for larger and longer-term studies to determine whether antifibrotic effects translate into clinically meaningful outcomes.
Sodium–glucose cotransporter 2 inhibitors (SGLT2i) have emerged as a cornerstone therapy in heart failure across a broad range of phenotypes. The believed mechanism for the cardioprotective effects of SGLT2i is the improvement of cardiac energetics, and their use in nonobstructive HCM has also been evaluated. In a smaller (n=48) prospective, open-label study involving patients with type 2 diabetes and symptomatic nonobstructive HCM with preserved left ventricular systolic function, treatment with SGLT2i over six months resulted in significant improvements in diastolic function parameters, a 6-minute walk test, and natriuretic peptide levels [29]. Additionally, SGLT2i use was associated with lower mortality, fewer hospitalisations, and improved symptoms without increased adverse events in a large observational HCM cohort [30]. Taken together, early data indicate that SGLT2 inhibitors may represent a promising adjunctive therapy in selected patients with HCM. Randomised controlled trials to assess both SGLT1 and SGLT2 inhibitors in patients with symptomatic obstructive and nonobstructive HCM are currently ongoing and upcoming [31,32].
Gene-targeted therapies
Though not yet approved, research in HCM is moving toward therapies that target the disease at the genetic level [33]. Because most pathogenic variants in HCM involve MYH7 and MYBPC3, emerging genetic therapies have primarily targeted these genes [34]. Several strategies, including gene or base editing, allele-specific silencing, and gene replacement therapies, are under investigation, but all are in pre-clinical or early clinical phases. Among these is a phase Ib/II study of TN-201, a gene therapy delivered using an adeno-associated virus vector, in HCM caused by MYBPC3 variants, including both obstructive and nonobstructive forms. Early results from the first three treated patients showed successful delivery of the therapy to cardiac tissue, increased levels of MYBPC3 protein expression, and good tolerability [35]. In parallel, gene editing and gene silencing approaches have reduced disease expression and myocardial hypertrophy in animal models, highlighting the potential of precision genetic therapies for selected HCM populations [33,35]. Together, these approaches highlight the growing promise of genetic therapies in HCM.
Beyond monogenic sarcomeric disease, accumulating evidence suggests that HCM also has a polygenic component, with common genetic variants modulating disease expression and penetrance [36].
Patient-oriented message
When discussing new treatments for HCM, it may be helpful to explain that emerging therapies may not only improve symptoms but also act on the underlying changes. Patients should understand that treatment is tailored to their specific presentation of HCM and that newer options may offer benefits beyond traditional medicines. Clear communication about expected effects, monitoring, and therapeutic goals supports shared decision-making and helps guide the selection of the most appropriate treatment.
Conclusions
The pharmacologic landscape of HCM is undergoing a transformative shift. The introduction of cardiac myosin inhibitors represents a transition from symptomatic management toward mechanism-based therapy. In obstructive HCM, these agents are now incorporated into clinical guidelines, with practice evolving accordingly. For clinicians managing HCM, familiarity with these therapies, including their indications, monitoring requirements, and limitations, is essential. As additional treatments, including gene-based approaches, advance toward clinical translation, therapeutic strategies may extend beyond symptom relief toward disease modification. Looking ahead, combined approaches may further personalise care and potentially alter disease progression by addressing key pathophysiological features of HCM.


