

















FOCUS on Cardiac Amyloidosis
July 2025

Table of contents
Cardiac amyloidosis is a progressive disease caused by the extracellular deposition of amyloid fibrils in the heart. More frequent than previously believed, it contributes to many common clinical scenarios such as severe aortic stenosis, increased left ventricular wall thickness and heart failure with preserved ejection fraction.
Today, we know that the most frequent subtype of cardiac amyloidosis in clinical practice is transthyretin amyloidosis (ATTR-CA) which is caused by transthyretin deposition. This subtype has an acquired or wild type (ATTRwt-CA) and a hereditary (ATTRv-CA) form.
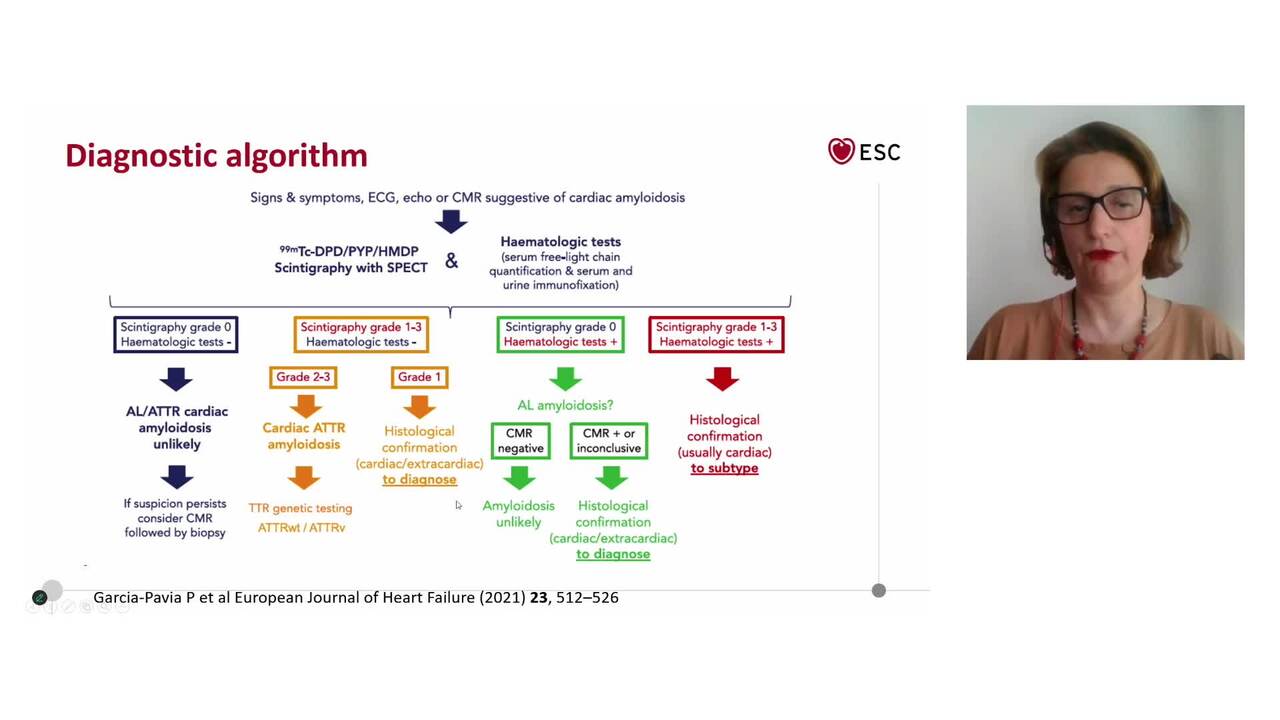
Most patients with ATTR-CA can be diagnosed without biopsies, simply by performing a scintigraphy and evaluating the presence of a monoclonal protein in the blood and urine.
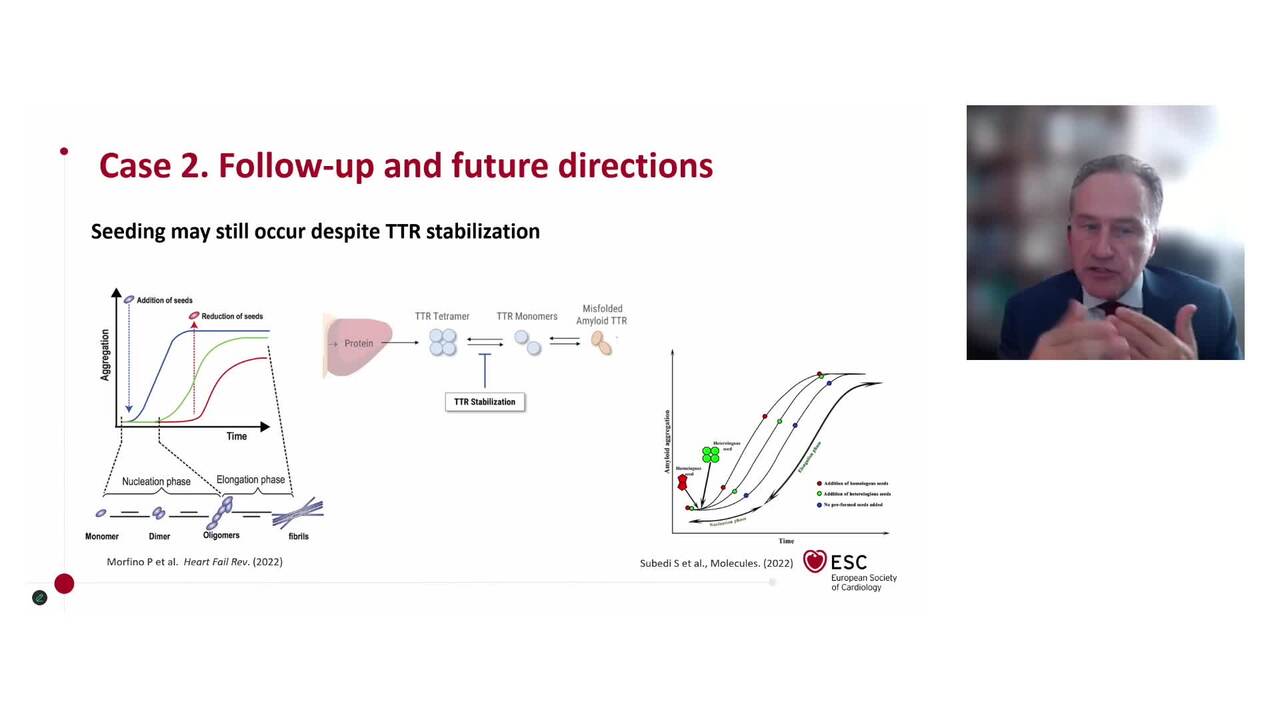
The prognosis of patients with ATTR-CA has improved in recent years, due to earlier diagnosis, closer follow-up and the development of specific therapies that target the amyloidogenic cascade.
This month’s FOCUS resources are all about cardiac amyloidosis. Build your knowledge about diagnosis, therapies and what we’ve learned from recent clinical trials.




Webinars
Webinar
When to suspect and how to diagnose cardiac amyloidosis (multimodality imaging)
7 Jul 2025 - 18:00
Webinar
New drugs in the treatment of amyloidosis
16 Jul 2025 - 18:00
Essentials


Relevant textbook chapters

Dig deeper
Exclusively for members
In practice
Webinar
Imaging in patients with HFpEF
2 Nov 2021 - 18:00
Webinar
Cardiac amyloidosis in valvular heart disease
29 Sep 2021 - 18:00
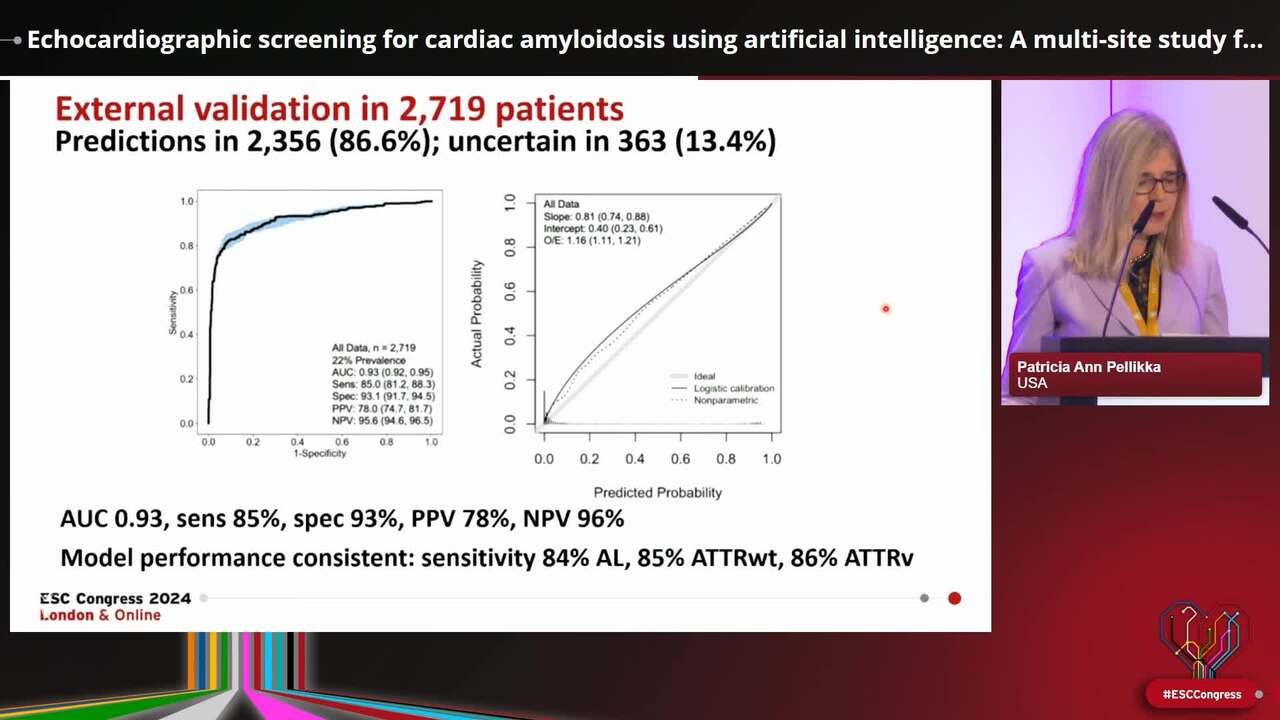
Congress Presentation
Echocardiographic screening for cardiac amyloidosis using artificial intelligence: A multi-site study for algorithm training and external validation.
1 Sep 2024 - 10:00
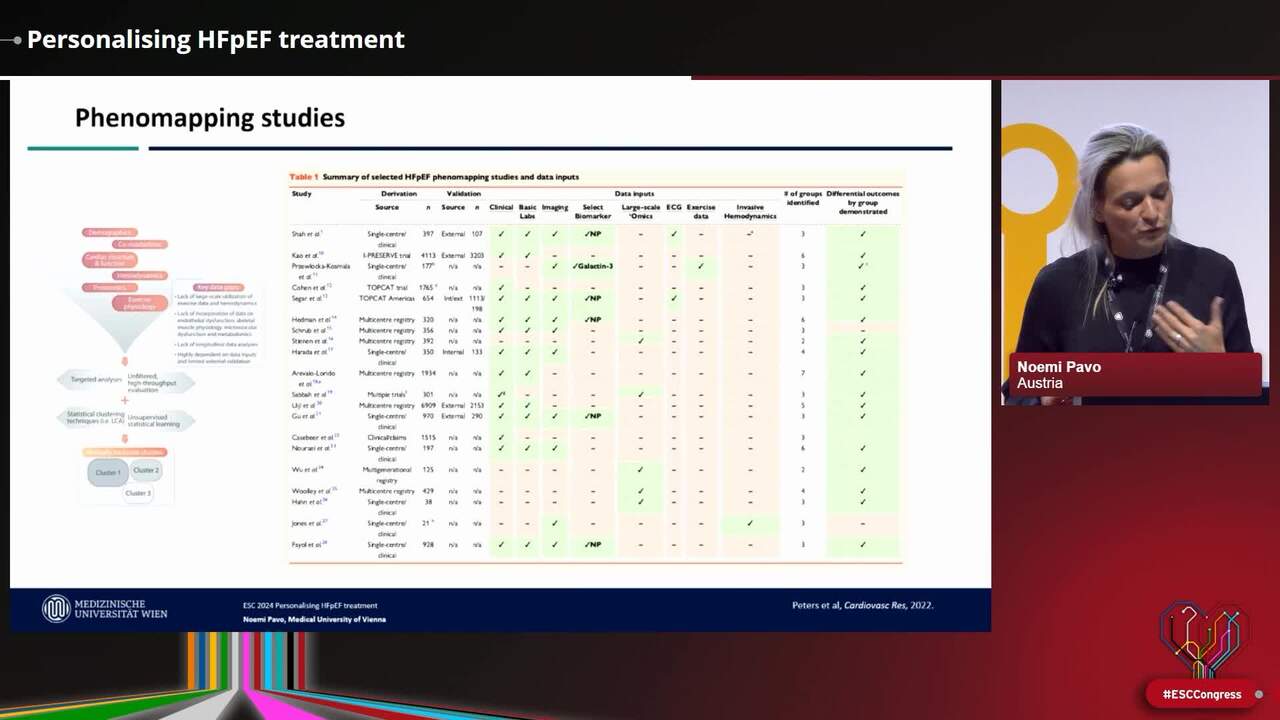
Congress Presentation
Personalising HFpEF treatment
1 Sep 2024 - 10:00

ESC TV Today
Episode 9: Management of cardiac amyloidosis - e-cigarettes and cardiovascular disease
15 Feb 2024 - 16:00
Interface with the specialists

Congress Presentation
Ask the Trialist - HELIOS-B
30 Aug 2024 - 14:45

Congress Presentation
HELIOS-B - Primary results from phase 3 study of vutrisiran in patients with transthyretin amyloidosis with cardiomyopathy
30 Aug 2024 - 12:00
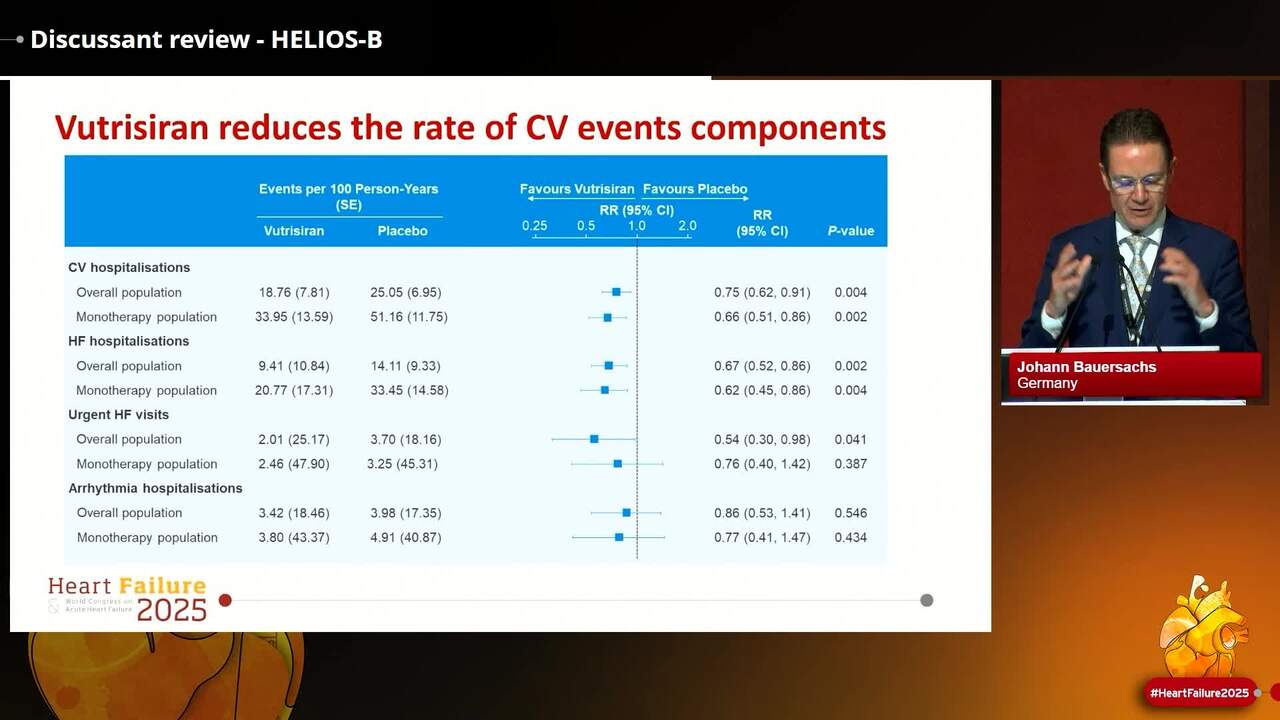
Congress Presentation
Discussant review - HELIOS-B
17 May 2025 - 11:58

Congress Presentation
HELIOS-B - Interview
30 Aug 2024 - 10:30
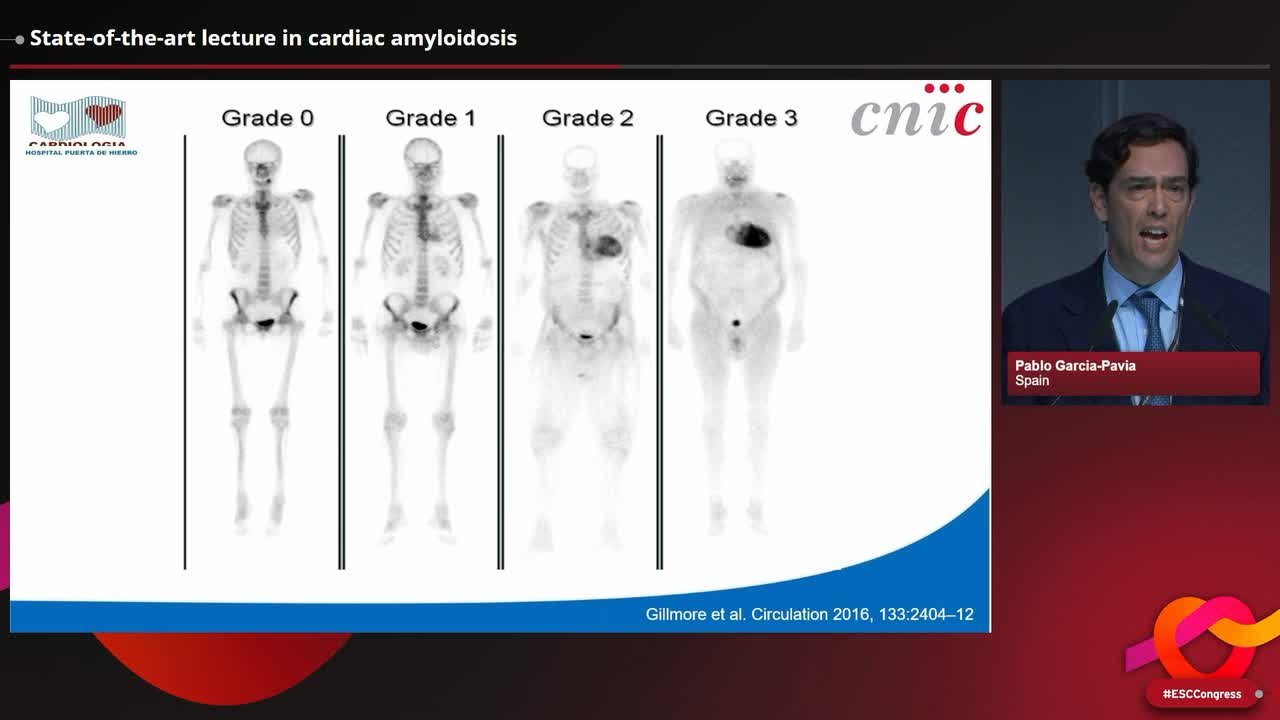
Congress Presentation
State-of-the-art lecture in cardiac amyloidosis
28 Aug 2022 - 14:00
Pioneers' Viewpoint

Podcast
Characteristics and natural history of early-stage cardiac transthyretin amyloidosis
13 Jun 2022 - 00:00
Congress Session
HELIOS-B - Interview
30 Aug 2024 - 10:30

European Heart Journal
Tafamidis for transthyretin amyloid cardiomyopathy: the solution or just the beginning of the end?
18 Jan 2019 - 00:00
This programme is supported by AstraZeneca through an independent support grant.
The scientific programme has not been influenced in any way by its sponsor.

ESC Pocket Guidelines Mobile App
Convenient resources available directly on your mobile device.
