Introduction
Early-onset atrial fibrillation (AF) can be the first manifestation of an underlying genetic predisposition to cardiomyopathy or arrhythmogenic syndromes. Genetic testing is increasingly recognized as a valuable tool for risk stratification and guiding patient management. Here, we present a case of a 40-year-old male with incidental AF detection and family history of sudden cardiac death, highlighting the role of genetic assessment in such cases.
Case Presentation
A 40-year-old man with no known prior medical history was found to have atrial fibrillation (AF) during a routine occupational health evaluation. The patient was asymptomatic at the time of detection and did not regularly engage in sports.
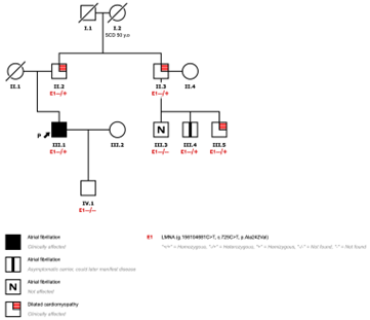
His family history was significant for a first-degree cousin with an unspecified heart disease and a maternal grandmother who died suddenly at age 50 at rest.
The patient was initially treated with bisoprolol 2,5 mg, anticoagulated with edoxaban 60 mg for three weeks, and scheduled for electrical cardioversion, which successfully restored sinus rhythm (Figure 1). Subsequently, pulmonary vein isolation (PVI) ablation was done. However, at 3 months follow-up and before PVI, the patient was found to be in a junctional escape rhythm secondary to sinus node dysfunction. He remained asymptomatic. Bisoprolol was discontinued, and a 24-hour Holter ECG confirmed a persistent junctional rhythm and no symptoms (Figure 2).
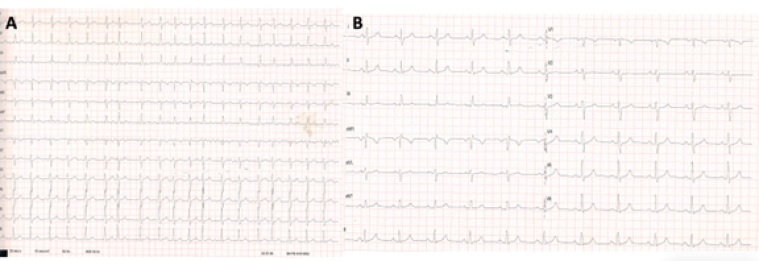
Figure 1 A) Atrial fibrillation with a ventricular rate of 120 bpm, normal electrical axis, and a narrow QRS complex. B) Sinus rhythm at 67 bpm, PR interval of 162 ms, QRS duration of 100 ms, with normal ST-segment and T-wave morphology.
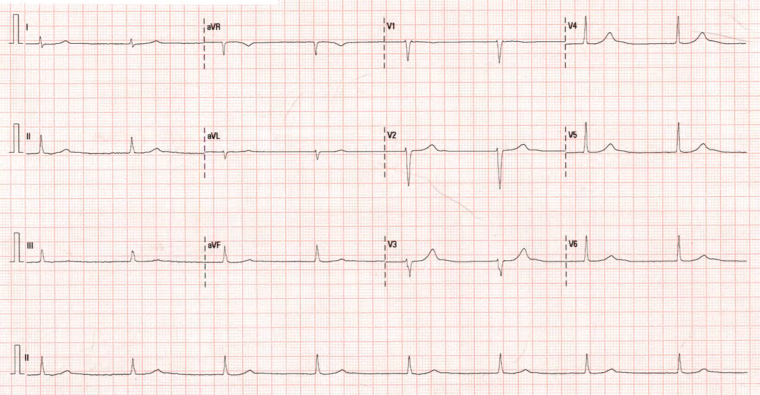
Figure 2: Junctional rhythm at 45 bpm, normal electrical axis, narrow QRS complex, and no repolarization abnormalities
An echocardiogram showed normal left ventricular size (end-diastolic diameter 52 mm), normal left ventricular ejection fraction (58%), maximal wall thickness of 11 mm, a non-dilated right ventricle with preserved systolic function (TAPSE 21 mm), and a mildly dilated left atrium (indexed left atrial volume of 30 mL/m²). Laboratory tests, including complete blood count, creatinine (0.9 mg/dL), potassium (4.2 mmol/L), creatine kinase (85 U/L), and NT-proBNP (110 pg/mL), were all within normal ranges. A cardiac MRI was performed, showing mid-myocardial late gadolinium enhancement in the basal-to-mid septal region, with normal left and right ventricular dimensions and systolic function. However, the left atrium was moderately dilated (indexed left atrial volume 42 mL/m²) (Figure 3).
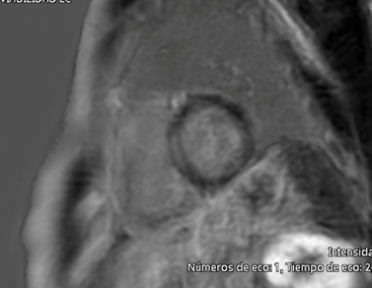
Figure 3: Cardiac Magnetic Resonance – Late gadolinium enhancement in the mid-myocardium of the interventricular septum.
Is there a link between sinus node disease and atrial fibrillation in this patient? Could this be an early sign of an underlying genetic cardiomyopathy? Should genetic testing be considered in this scenario?
The presence of atrial fibrillation in a young male, particularly in the absence of conventional risk factors, raises suspicion of an underlying cardiomyopathy or genetic predisposition. This concern is further heightened by a family history of sudden cardiac death and the evolution of additional rhythm disturbances, such as sinus node dysfunction. These findings suggest a potential inherited arrhythmogenic or structural myocardial disease. From a genetic perspective, several conditions could explain this phenotype. Variants frequently associated with NDLVC (LMNA, SCN5A, DES), ARVC (PKP2, JUP) dilated cardiomyopathy (TTN, MYH7, BAG3), and primary arrhythmia syndromes (KCNQ1, KCNH2, KCNJ2) should be considered. According to the evidence, the diagnostic yield of genetic testing in individuals with atrial fibrillation under 50 years of age is approximately 10–13%. The most frequently detected variants are found in the TTN gene. Although the ESC guidelines for atrial fibrillation don’t make specific recommendation for genetic testing, the American College of Cardiology/American Heart Association guidelines recommended genetic testing in AF patients aged 45 years or younger (Class IIb).
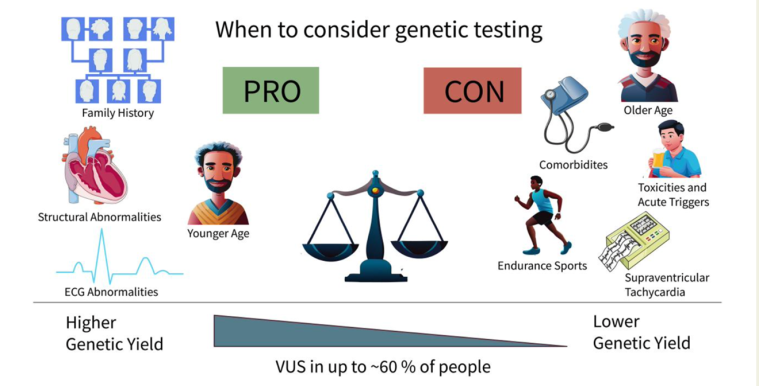
Figure 4. Overview of when to consider genetic testing and expected diagnostic yield in rare variant testing in early-onset atrial fibrillation across lifetime (Kany S et al Eur Heart J. 2024)
Given his clinical presentation and family history, pedigree analysis and genetic testing were performed using a 121-gene cardiomyopathy panel. This revealed a missense variant in the LMNA gene (NM_170707.3:c.725C>T, p.Ala242Val), classified as likely pathogenic. (Figure 4)
At the time of the scheduled exercise stress test, the patient was again found to be in AF, though with an adequate ventricular rate. The stress test was clinically and electrically non-conclusive, with no significant arrhythmic events. Due to recurrent AF episodes, a rate-control strategy was preferred over rhythm control. The genetic background and the size of left atrium led to taking this treatment. The patient was maintained on edoxaban, and enalapril was added to his treatment regimen to optimize cardiac remodeling and reduce progression to cardiomyopathy. In this patient, genetic testing mandates reassessment of the risk of ventricular arrhythmias and sudden cardiac death. The LMNA-risk VTA calculator showed 6,6% risk of Life-Threatening Ventricular Tachyarrhythmias at 5 years. We decided on medical management, clinical follow-up and close monitoring.
The LMNA p.Ala242Val variant is associated with “laminopathies”, which include a spectrum of cardiac and systemic manifestations such as dilated cardiomyopathy (DCM), conduction system disease, and arrhythmias. LMNA mutations are known to carry a high risk for progressive conduction disease, malignant ventricular arrhythmias, and sudden cardiac death, often necessitating close monitoring and, in some cases, early consideration of an implantable cardioverter-defibrillator (ICD).
This case underscores the importance of genetic testing in early-onset AF, especially in patients with suggestive family history and additional cardiac findings such as left atrial enlargement or conduction abnormalities. The presence of an LMNA mutation in an AF patient raises concern for the potential development of cardiomyopathy and/or life-threatening arrhythmias, necessitating long-term follow-up and family screening.