Methods and Results:
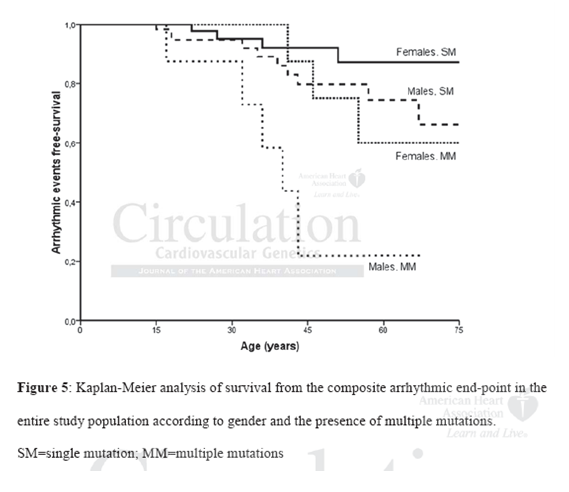
The study population consisted of 134 carriers of mutations in genes encoding desmosomal proteins (44 probands, 90 family members). Endpoint outcome was defined as sudden cardiac death (SCD), aborted SCD, sustained ventricular tachycardia and appropriated ICD intervention.113 (84%) individuals had a single mutation which in DSP (n=44), PKP-2 (n=38), DSG-2 (n=30) and DSC-2 (n=1); in 62 of them its was a missense mutation and in the rest 51 a non-missense mutation. 21 (16%) individuals had complex mutations; compound heterozygocity was present in 7 and digenic heterozygocity in 14. Disease penetrance, defined as either a definite or borderline diagnosis based on the 2010 revised task force criteria, occurred in 78 (58%) mutation carriers. Carriers of multiple mutations had higher rates of disease penetrance (p=0.03). Among these patients low voltage and left ventricular (LV) dysfunction was more frequent within males (p values 0.04 and 0.001 respectively). Multiple mutation carriers exhibited an increased incidence of epsilon waves (p=0.001), right precordial T-wave inversion (TWI) beyond V3 (p=0.02), late potentials (p=0.04), lower right ventricular functional (RV) area change (p=0.001) and multiple right ventricular wall motion abnormalities (p=0.01). Within the group of single mutation carriers, increased incidence of right precordial TWI beyond lead V3 (p=0.006) and lower RV fractional area change (p=0.06) was found in non-missense as compared to missense mutation carriers. Individuals with a single mutation in the DSP gene had more frequent TWI in leads V4 to V6 (p=0.02), low QRS voltage in limb leads (p=0.02) and LV dysfunction (p=0.01) as compared to single mutation carriers of the rest desmosomal genes. 22 (16%) of the mutation carriers reached the endpoint outcome (12/44 probands and 10/90 family members). Multivariate analysis revealed the presence of multiple mutations and male gender to be independent predictors of the endpoint outcome (HR 3.71 and 2.76 respectively). Lifetime endpoint rate was incremental between the groups of females with single mutation (7%), males with single mutation (13%), females with multiple mutations (27%) and males with multiple mutations (63%).
Discussion:
The main result of this study is that the presence of a complex genotype was found in 16% of the cohort and independently predicted the occurrence of major arrhythmic events. Although, this hypothesis has been suggested before, this is the first study capable to support it. Additionally this group of patients experienced an increased disease penetrance with more prevalent depolarization and repolarization changes on ECG and structural/functional alterations on two-dimensional echocardiography. These findings suggest that multiple mutations probably have an additive effect on disease pathophysiology and that comprehensive screening of all desmosomal genes might be beneficial for patients in terms of risk assessment.
The study population consisted of 134 carriers of mutations in genes encoding desmosomal proteins (44 probands, 90 family members). Endpoint outcome was defined as sudden cardiac death (SCD), aborted SCD, sustained ventricular tachycardia and appropriated ICD intervention.113 (84%) individuals had a single mutation which in DSP (n=44), PKP-2 (n=38), DSG-2 (n=30) and DSC-2 (n=1); in 62 of them its was a missense mutation and in the rest 51 a non-missense mutation. 21 (16%) individuals had complex mutations; compound heterozygocity was present in 7 and digenic heterozygocity in 14. Disease penetrance, defined as either a definite or borderline diagnosis based on the 2010 revised task force criteria, occurred in 78 (58%) mutation carriers. Carriers of multiple mutations had higher rates of disease penetrance (p=0.03). Among these patients low voltage and left ventricular (LV) dysfunction was more frequent within males (p values 0.04 and 0.001 respectively). Multiple mutation carriers exhibited an increased incidence of epsilon waves (p=0.001), right precordial T-wave inversion (TWI) beyond V3 (p=0.02), late potentials (p=0.04), lower right ventricular functional (RV) area change (p=0.001) and multiple right ventricular wall motion abnormalities (p=0.01). Within the group of single mutation carriers, increased incidence of right precordial TWI beyond lead V3 (p=0.006) and lower RV fractional area change (p=0.06) was found in non-missense as compared to missense mutation carriers. Individuals with a single mutation in the DSP gene had more frequent TWI in leads V4 to V6 (p=0.02), low QRS voltage in limb leads (p=0.02) and LV dysfunction (p=0.01) as compared to single mutation carriers of the rest desmosomal genes. 22 (16%) of the mutation carriers reached the endpoint outcome (12/44 probands and 10/90 family members). Multivariate analysis revealed the presence of multiple mutations and male gender to be independent predictors of the endpoint outcome (HR 3.71 and 2.76 respectively). Lifetime endpoint rate was incremental between the groups of females with single mutation (7%), males with single mutation (13%), females with multiple mutations (27%) and males with multiple mutations (63%).
Discussion:
The main result of this study is that the presence of a complex genotype was found in 16% of the cohort and independently predicted the occurrence of major arrhythmic events. Although, this hypothesis has been suggested before, this is the first study capable to support it. Additionally this group of patients experienced an increased disease penetrance with more prevalent depolarization and repolarization changes on ECG and structural/functional alterations on two-dimensional echocardiography. These findings suggest that multiple mutations probably have an additive effect on disease pathophysiology and that comprehensive screening of all desmosomal genes might be beneficial for patients in terms of risk assessment.
Conclusion:
However it is, yet, unclear how this information can be implemented into a more comprehensive clinical risk stratification model for patients with ARVC. Under these circumstances, in the era of next generation sequencing, observed genetic variances need to be interpreted with great care in order not to mislead patient diagnosis and management.