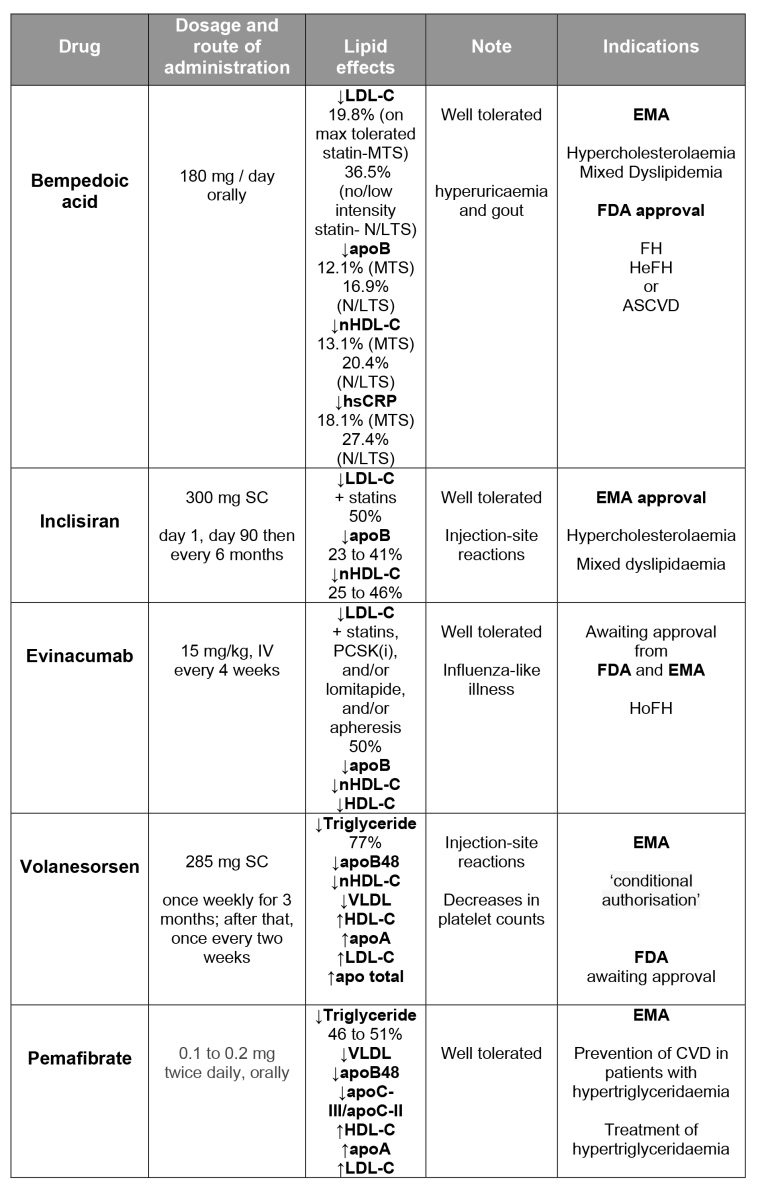
Bempedoic acid
Bempedoic acid (BA) is an oral, once-daily, first-in-class, small-molecule cholesterol synthesis inhibitor for the treatment of hyperlipidaemia. As an adenosine triphosphate (ATP)-citrate lyase inhibitor, BA acts upstream of HMG-CoA reductase to inhibit cholesterol biosynthesis and increases LDL receptor expression. BA is a prodrug, which only becomes activated in the hepatocyte; therefore, potential muscle adverse effects, which are mostly associated with statins, were not observed [1].
Current clinical status of BA
In a pooled analysis of 3,623 patients with hypercholesterolaemia (4 randomised clinical trials of a BA clinical programme), the results showed a significant reduction of low-density lipoprotein cholesterol (LDL-C), ranging between 17 and 25% versus placebo (depending on statin background therapy). Significant improvements were also seen in total cholesterol non-high-density lipoprotein cholesterol (non-HDL-C), apolipoprotein B (apoB) and high-sensitivity C-reactive protein (hsCRP) levels. These improvements in lipid parameters were maintained throughout the treatment period and were consistent across different patient subgroups (background lipid-lowering therapy [LLT]; primary vs secondary prevention; other comorbidities) [2]. In order to describe the safety profile of BA treatment, a second pooled analysis of data from the four phase 3 RCTs was performed. BA was well tolerated with no major safety concerns. In this pooled analysis, new-onset diabetes and worsening of diabetes occurred less frequently with BA versus placebo. Background LLT had no demonstrable effect on the safety or tolerability of BA versus placebo [3].
In a phase 3 clinical trial, SERENITY, in patients with a history of statin intolerance and requiring additional LDL-C lowering, BA 180 mg once daily was effective in reducing LDL-C when added to ezetimibe 10 mg per day with or without additional lipid-modifying therapy (LMT). There was an additional reduction of 23.6% in LDL-C compared with placebo and there was also a reduction in the non-HDL-C, total cholesterol, and apoB of 19.0%, 16.1% and 15.5%, respectively. Additionally, hsCRP was also reduced by 25.4%. BA was safe and well tolerated, without any increase of side effects such as myalgia or muscular weakness [4]. In patients with high/very high cardiovascular risk and elevated LDL-C not adequately controlled with maximally tolerated statin either alone or in combination with other LMT, a fixed-dose combination of BA and ezetimibe (180 mg and 10 mg, respectively) significantly lowered LDL-C versus placebo (–36.2% versus 1.8% from placebo; p<0.001). Moreover, a favourable safety profile was also observed [5]. In a meta-analysis including 11 RCTs of BA, enrolling 4,391 patients, its use was associated with reductions in LDL-C and hsCRP levels, composite CV outcomes and a reduction in rates of new-onset or worsening diabetes [6].
The effects of BA on cardiovascular (CV) morbidity and mortality have not been studied; the results of the Evaluation of Major Cardiovascular Events in Patients With, or at High Risk for, Cardiovascular Disease Who Are Statin Intolerant Treated With Bempedoic Acid (ETC-1002) or Placebo (CLEAR Outcomes) are awaited [7]. The study randomised 14,014 patients to treatment with BA 180 mg daily or matching placebo on a background of guideline-directed medical therapy. The primary outcome is a composite of the time to first CV death, non-fatal myocardial infarction, non-fatal stroke or coronary revascularisation.
The U.S. Food and Drug Administration (FDA) has approved BA for the treatment of adults with heterozygous familial hypercholesterolaemia (HeFH) or established atherosclerotic CV disease and who require additional lowering of LDL-C, and the European Medicines Agency (EMA) decided that BA benefits are greater than its risks and that it can be authorised for use in the European Union (EU).
Inclisiran
Inclisiran, a small interfering ribonucleic acid (RNA) targeting hepatic PCSK-9 synthesis, has the potential to reduce LDL-C levels substantially with an acceptable side-effect profile and an infrequent dosing regimen; it is administered subcutaneously with an initial dose, repeated at 3 months and then every 6 months.
Current clinical status of inclisiran
Men and women aged 18 or over, with an LDL-C higher than 70 or 100 mg/dl (with or without a history of atherosclerotic CV disease) and receiving the maximum possible dose of a statin with or without additional LMT, were administered a single dose of placebo or 200, 300, or 500 mg of inclisiran on day 1 or two doses of placebo or 100, 200, or 300 mg of inclisiran on day 1 and day 90 (ORION-1). Inclisiran treatment resulted in a significantly greater reduction in LDL-C levels at 180 days than in the group of patients receiving placebo. The greatest reduction (52.6%) in LDL-C was observed in association with the two-dose 300 mg regimen, a reduction that is in a range similar to that achieved with PCSK-9 antibodies.
The most common adverse events (>2% of patients) were myalgia, headache, fatigue, nasopharyngitis, back pain, hypertension, diarrhoea and dizziness; the incidences of these events did not differ significantly between inclisiran and placebo. Injection-site reactions occurred in 5% of the inclisiran patient group [8]. In the phase 3 trial in adults with HeFH, receiving a maximally accepted dose of statin therapy with or without ezetimibe, those who received inclisiran (at a dose of 300 mg), administered as a subcutaneous injection on days 1, 90, 270 and 450, had significantly lower levels of LDL-C than those who received placebo. The reduction was 39.7% in the inclisiran group, while an increase of 8.2% was observed in the placebo group, accounting for a total difference of −47.9% (p<0.001). Again, the adverse events and serious adverse events were similar in the two groups [9].
Patients with atherosclerotic cardiovascular disease (ASCVD) (ORION-10 trial) and patients with ASCVD or an equivalent ASCVD risk (ORION-11 trial), who had elevated LDL-C levels despite receiving statin therapy at the maximum tolerated dose (>70 mg/dl for patients with ASCVD and >100 mg/dl for patients with an ASCVD equivalent), were randomly assigned in a 1:1 ratio to receive either inclisiran (284 mg) or placebo. Reductions in LDL-C levels were approximately 50% in the group receiving inclisiran, administered subcutaneously every 6 months [10]. The results from ORION-4, which enrolled 15,000 participants (with at least one of the following conditions: prior myocardial infarction, or prior ischaemic stroke, or peripheral artery disease, or aortic aneurysm repair), are awaited. The primary outcome was defined as the time to first event occurrence - coronary heart disease death, myocardial infarction, fatal or non-fatal ischaemic stroke or urgent coronary revascularisation procedure [11].
The adverse effects of inclisiran were reported to be mild or moderate, with the most common ones occurring with similar frequency either in the inclisiran group or in the placebo group. Laboratory results with respect to liver and kidney function, levels of creatine kinase and hsCRP and platelet count were also similar in the inclisiran and placebo groups. Injection-site adverse events were more frequent with inclisiran than with placebo [10].
The Committee for Medicinal Products for Human Use (CHMP) of the EMA has adopted a positive opinion and recommended granting marketing authorisation of inclisiran for the treatment of adults with hypercholesterolaemia or mixed dyslipidaemia.
Evinacumab
Evinacumab is a fully human monoclonal antibody that is an inhibitor of angiopoietin-like 3 (ANGPTL3). ANGPTL3 is an inhibitor of lipoprotein and endothelial lipase. It plays a key role in lipid metabolism by increasing the levels of triglycerides and other lipids [12]. Loss-of-function variants in ANGPTL3 have been associated with low levels of both LDL-C and triglycerides and with a 41% lower risk of coronary artery disease, despite the presence of low levels of HDL-C [13, 14].
Current clinical status of evinacumab
The Efficacy and Safety of Evinacumab in Patients With Homozygous Familial Hypercholesterolemia (ELIPSE HoFH) trial is a phase 3, randomised, placebo-controlled, parallel-group trial, where the efficacy and safety of evinacumab (administered intravenously every four weeks) was evaluated in 65 patients with null-null variants and in those with non-null variants of LDL-receptor activity. The mean baseline LDL-C level was 260 mg per decilitre in the evinacumab group and 247 mg per decilitre in the placebo group. At baseline, the mean LDL-C level was 312±158 mg per decilitre in the patients with null–null variants and 228±164 mg per decilitre in the 44 patients with non-null variants. The majority of the trial patients (94%) were receiving a statin (a high-intensity statin in 77%). In addition, a PCSK-9 inhibitor was being administered in 77% of the patients, ezetimibe in 75% and lomitapide in 25%, and 34% of the patients were undergoing apheresis. A total of 63% of the patients were taking at least three LMT drugs. At week 24, patients in the evinacumab group had a 47.1% reduction from baseline in the LDL-C level, as compared with a 1.9% increase in the placebo group, for a between-group mean difference of –49.0% (p<0.001). The degree of cholesterol-lowering levels was verified among patients with null–null variants (–43.4% and +16.2%, respectively) and non-null variants (–49.1% and –3.8%, respectively). Patients in the evinacumab group also had significantly lower levels of apoB, non-HDL-C, HDL-C and total cholesterol from baseline at week 24 as compared with those in the placebo group (p<0.001).
Evinacumab was generally well tolerated. None of the patients discontinued the drug because of an adverse event. The most common adverse event reported was an influenza-like illness, observed in 11% of the patients in the evinacumab group [15].
The FDA has accepted for Priority Review a Biologics License Application for evinacumab as an adjunct to other LMT in patients with HoFH, and the CHMP has recommended an accelerated assessment for evinacumab.
Volanesorsen
Volanesorsen is an antisense oligonucleotide that binds to apoC-III mRNA, leading to its degradation, and preventing translation of apoC-III protein, which inhibits triglyceride metabolism and hepatic clearance of chylomicrons. Preventing translation of apoC-III allows metabolism and breakdown of these triglycerides and chylomicrons [16]. It is administered subcutaneously, once weekly for 3 months. Following 3 months, the dose frequency is reduced to once every two weeks.
Current clinical status of volanesorsen
Several clinical trials have documented the efficacy of volanesorsen in the treatment of familial chylomicronaemia syndrome (FCS) and elevated triglycerides (TGs). The APPROACH trial was a randomised, double-blind, phase 3 trial that evaluated the efficacy and safety of volanesorsen (at a dose of 300 mg subcutaneously once a week) compared with placebo in adult patients who had FCS. Patients were eligible to participate if the syndrome was confirmed either by genetic testing or by documentation of low lipoprotein lipase (LPL) activity and if fasting triglyceride levels were at or above 750 mg/dl. Volanesorsen reduced mean apoC-III levels from baseline by 84% after 3 months of therapy and by 83% after 6 months of therapy (p<0.001); in contrast, mean apoC-III levels increased by 6.1% after 3 months of therapy and decreased by 5.2% at 6 months with placebo.
The mean percentage change in triglyceride levels between baseline and 3 months was a decrease of 77% in the volanesorsen group and an 18% increase in the placebo group (p<0.001), which corresponded to a mean decrease of 1,712 mg/dl in the volanesorsen group and an increase of 92.0 mg/dl in the placebo group (p<0.001). Volanesorsen also decreases the levels of the apoB48 (76%; p<0.001), non-HDL-C (46%; p< 0.001) and very low-density lipoprotein (VLDL) (58%; p<0.001), and increases the levels of HDL-C (46%; p<0,001), apoA1 (14%; p<0.001), LDL-C (136%; p<0.001) and total apoB (20%; p=0.03).
The most common adverse events that occurred during treatment among patients who received volanesorsen were those related to injection-site reactions and decreases in platelet counts. A platelet count below 140,000 per microlitre was observed in 76% in the volanesorsen group and in 24% in the placebo group; levels below 100,000 per microlitre were observed in 48% of patients who received volanesorsen, but not in patients who received placebo. Thrombocytopaenia required more frequent monitoring and an adjustment in the frequency of injections [17]. Results from the pooled analysis of both the COMPASS trial (114 participants with fasting TG ≥500 mg/dL randomised 2:1 to volanesorsen or placebo) and the APPROACH trial showed a significant reduction (p=0.0185) in pancreatitis [18].
Volanesorsen has been given “conditional authorisation” by the EMA, meaning that, for patients at high risk of pancreatitis and for whom other medicines and a low-fat diet have not worked well enough, the benefits of volanesorsen are greater than its risks.
Pemafibrate
Pemafibrate is a novel selective PPARα modulator with higher potency and selectivity for PPARα activation than fenofibrate. The available preclinical and clinical data suggest that this new drug, due to its favourable benefit/risk balance, is more effective than conventional PPARα agonists.
Current clinical status of pemafibrate
In a phase 3, placebo/active drug-controlled, double-blind, parallel group comparison study that enrolled patients with high TG and low HDL-C levels, the participants were randomly assigned to receive placebo or pemafibrate 0.1 mg/day, 0.2 mg/day, or 0.4 mg/day or fenofibrate 100 mg/day or 200 mg/day during 12 weeks. Pemafibrate 0.2 mg/day and 0.4 mg/day were confirmed to be non-inferior to fenofibrate 200 mg/day, and pemafibrate 0.1 to 0.4 mg/day was confirmed to be superior to fenofibrate 100 mg/day for the reduction of TG levels. The incidence of adverse events and/or adverse drug reactions in all pemafibrate groups was confirmed to be comparable to that of placebo and less frequent than that of fenofibrate 200 mg/day. Overall, pemafibrate 0.4 mg/day was superior to fenofibrate 200 mg/day [19].
The PROMINENT trial is an ongoing trial with approximately 10,000 participants randomised to either pemafibrate (0.2 mg twice daily) or matching placebo, with an average expected follow-up period of 3.75 years. The elected patients have type 2 (T2) diabetes, hypertriglyceridaemia (TG: 200-499 mg/dl) and low HDL levels (≤40 mg/dl). All patients must be receiving either moderate-to-high intensity statin therapy or meet specified LDL-C criteria. The study includes patients in primary and secondary prevention. The primary endpoint is a composite of non-fatal myocardial infarction, non-fatal ischaemic stroke, hospitalisation for unstable angina requiring urgent coronary revascularisation and cardiovascular death [20].
The EMA decided that pemafibrate has a role in the treatment of patients with hypertriglyceridaemia and in the prevention of CV events in patients with hypertriglyceridaemia.
Conclusion
In the last few years, we have witnessed the emergence of new lipid-lowering drugs. Table 1 below shows the main characteristics of these new drugs. Some of them have a mechanism of action already explored, such as the cholesterol synthesis inhibition by bempedoic acid and the PPARα activation such as pemafibrate. Others are exploring new technologies, such as the use of monoclonal antibodies (evinacumab) and antisense oligonucleotides (inclisiran and volanesorsen). Bearing in mind the ambitious therapeutic targets recommended by the new ESC guidelines [21], combined therapy using the oldest and these new drugs permits increasing attainment of the desired and needed lipid goals.