

















Abbreviations
Cpc-PH: combined post-capillary pulmonary hypertension
CTEPH: chronic thromboembolic pulmonary hypertension
DPG: diastolic pressure gradient
ESC/ERS: European Society of Cardiology/European Respiratory Society
FAC: fractional area change
iPAH: idiopathic pulmonary arterial hypertension
Ipc-PH: isolated post-capillary pulmonary hypertension
LV: left ventricle
NO: nitric oxide
PAH: pulmonary arterial hypertension
PAPm: mean pulmonary arterial pressure
PAWP: pulmonary artery wedge pressure
PH: pulmonary hypertension
RV: right ventricle
S ́: systolic excursion velocity of the tricuspid annulus
TAPSE: tricuspid annular plane systolic excursion
TR: tricuspid regurgitation
WU: Wood units
Keywords
drug therapy, exercise capacity, prognosis, pulmonary hypertension, right heart failure
Background
Pulmonary hypertension (PH) is defined as an elevated mean pulmonary arterial pressure (PAPm) of ≥25 mmHg at rest as assessed by right heart catheterisation according to the 2015 ESC/ERS Guidelines [1]. A great variety of clinical conditions is connected causally to this heterogeneous disease. The clinical classification of PH differentiates roughly between pre-capillary PH (pulmonary artery wedge pressure [PAWP] ≤15 mmHg) and post-capillary PH (PAWP >15 mmHg).
The classification of pre-capillary PH includes:-
- idiopathic, heritable, and associated forms (Group 1, pulmonary arterial hypertension [PAH])
- PH due to lung diseases and/or hypoxia (Group 3)
- Chronic thromboembolic PH (CTEPH) and other pulmonary artery obstructions (Group 4)
- PH with unclear and/or multifactorial mechanisms (Group 5)
The classification of post-capillary PH (due to left heart disease Group 2, and in part Group 5) includes:-
- Isolated post-capillary PH (Ipc-PH) with diastolic pressure gradient (DPG) <7 mmHg and/or pulmonary vascular resistance (PVR) ≤3 Wood units (WU)
- Combined post-capillary PH (Cpc-PH) with DPG ≥7 mmHg and/or PVR >3 WU
Progressive pulmonary vascular remodelling is a common hallmark of PH, with the degree of vasculopathy varying according to the PH type. Thickening of the vascular wall, in situ thrombosis, and narrowing of the vascular lumen occurs particularly in PAH [2]. Exposed to a continuously increasing afterload, the thin-walled right ventricle (RV) - able to handle a low resistance circulation - must adapt to pressure and volume overload, whereupon the latter is better tolerated in general [3]. The increased wall stress leads either to concentric remodelling of the RV with preserved function (adaptive remodelling) or to maladaptive remodelling, which is characterised by eccentric hypertrophy and worse function [4]. Fluid retention and low cardiac output leading to exercise intolerance, fatigue, and arrhythmias are characteristics of the resulting complex clinical syndrome, designated RV failure. The most common causes of RV failure are chronic left heart failure and PH [3,5].
Prognostic importance of PH
Depending on the PH aetiology, i.e., the clinical classification as described above, the prognosis of patients with PH varies widely. Group 1 PH (PAH) is in general a progressive, “malignant” disease with a very poor prognosis of up to <70% one-year survival after first diagnosis [6]. In PAH patients, risk prediction is based on a relatively solid database: functional capacity, biomarkers, imaging and haemodynamics are used as predictors of survival and for classification of patients into low, intermediate, and high risk [7,8]. More or less all parameters used reflect the extent of RV dysfunction, which is crucial for the patients’ prognosis in all PH groups, independent of the aetiology [9,10]. The definition of RV dysfunction is much less established than the definition of left ventricular (LV) systolic dysfunction, where the use of the echocardiographically determined LV ejection fraction is generally accepted as a reasonable compromise. There is a multitude of echocardiographic parameters used to determine RV function but no broad consensus about which of them should be used routinely. There is only agreement that several parameters should be used, for example the combination of RV fractional area change (FAC), systolic excursion velocity of the tricuspid annulus (S ́), and tricuspid annular plane systolic excursion (TAPSE) [11]. Multiple morphologic and haemodynamic parameters characterise RV dysfunction, whereby MRI-derived RV ejection fraction is considered as the gold standard for describing RV function and haemodynamic parameters are considered to be standard indices of outcome in PAH [8,12]. Prognostic relevance, however, is limited to the few parameters derived from right heart catheterisation. For example, therapy-related lowering of elevated pulmonary vascular resistance (which is a fixed component of the PAH definition) does not automatically mean improvement of RV dysfunction and prognosis [13]. An overview of parameters that are prognostically relevant in terms of therapy goals for PAH patients is shown in Table 1.
Table 1. Risk assessment in pulmonary arterial hypertension. The goal of therapy is to lower risk by improvement of single parameters.
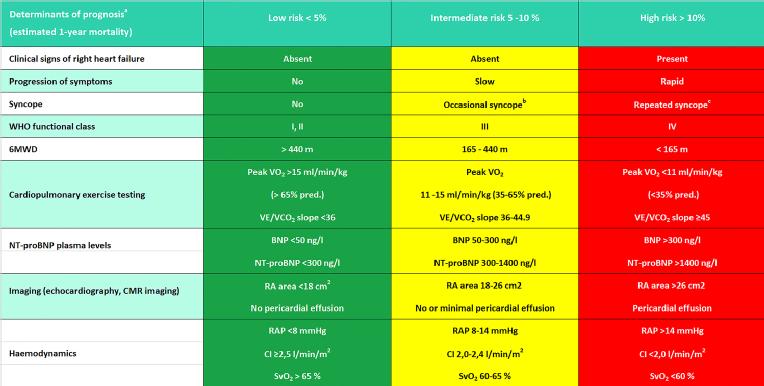
6MWD = 6-minute walking distance; BNP = brain natriuretic peptide; CI = cardiac index; CMR = cardiac magnetic resonance; NT-proBNP = N-terminal pro-brain natriuretic peptide; pred. = predicted; RA = right atrium; RAP = right atrial pressure; SvO2 = mixed venous oxygen saturation; VE/VCO2 = ventilatory equivalents for carbon dioxide; VO2 = oxygen consumption; WHO = World Health Organization.
aMost of the proposed variables and cut-off values are based on expert opinion. They may provide prognostic information and may be used to guide therapeutic decisions, but application to individual patients must be done carefully. One must also note that most of these variables have been validated mostly for iPAH and the cut-off levels used above may not necessarily apply to other forms of PAH. Furthermore, the use of approved therapies and their influence on the variables should be considered in the evaluation of the risk.
bOccasional syncope during brisk or heavy exercise, or occasional orthostatic syncope in an otherwise stable patient.
cRepeated episodes of syncope, even with little or regular physical activity.
2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC),International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016 Jan 1;37(1):67-119. http://eurheartj.oxfordjournals.org/content/37/1/67.
By permission of Oxford University Press
Treatment of PH
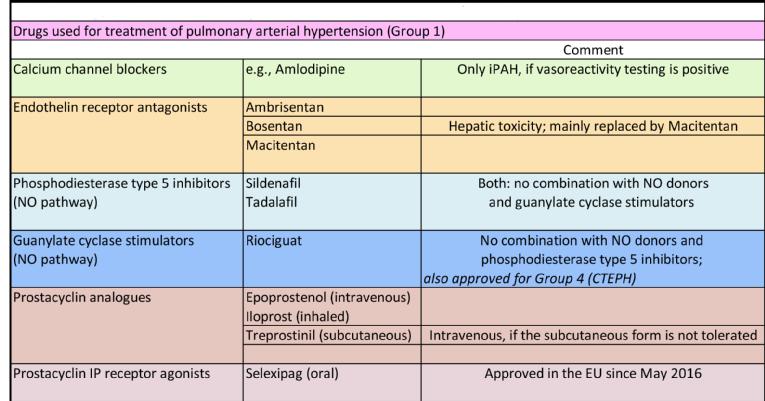
Approved therapies exist for PAH (Group 1) and CTEPH (Group 4). Drug therapy targets the three main pathways involved in the pathogenesis of PAH: prostacyclin, endothelin, and nitric oxide (NO) pathways. Early or initial combination therapy with agents targeting more than one pathogenic pathway is a favoured approach in patients with severe haemodynamic compromise [5]. An essential effect of drugs acting as agonists (prostacyclin, NO) or antagonists (endothelin) of these pathways is vasodilation; other effects such as anti-inflammation and antiproliferation contribute less to the treatment of the disease. Treatment with high-dose calcium channel blockers is reserved for patients with idiopathic PAH showing vasoresponsiveness in a so-called vasoreactivity testing procedure. Table 2 gives an overview of drugs approved for PAH.
Treatment can result in an improvement of exercise tolerance and quality of life, a delay in disease progression, and an improvement of prognosis; the one-year mortality, however, remains about 15% [5,8,14]. The only drug approved for use with patients outside Group 1 is riociguat, a stimulator of the NO pathway, which is also approved for Group 4. Importantly, the primary therapy of CTEPH is surgical (pulmonary endarterectomy), and drug therapy is considered as an alternative only if surgery is not possible. If surgical or drug treatment is successful, ameliorated RV function can lead - seemingly paradoxically - to higher pulmonary pressures, especially during exercise [15]. In end-stage PAH, the RV dilates and fails progressively, and pulmonary pressures drop in parallel with cardiac output while right atrial pressure rises [8]. In some selected patients, lung transplantation or balloon atrial septostomy may be considered.
For Group 2, 3, and 5 PH patients, there are no approved specific therapies available beyond treatment of the underlying disease. In particular, this is a major dilemma for Group 2 (PH due to left heart disease) patients because of the high frequency of its occurrence and the commonly adverse course of the disease. When causal therapy is exhausted, treatment of LV failure with a mechanical device is highly effective; however, ventricular assist devices are indicated only rarely, for example as a treatment of severe pulmonary hypertension to enable candidacy for heart transplantation.
Whereas several small studies have shown certain improvements by treatment of PH patients with left heart disease with drugs approved for PAH, some larger randomised trials failed to reach their endpoints [5]. The basic problem seems to be the uncertainties of the pathophysiology of the disease and a lack of promising treatment goals in these patients; the latest data from the European COMPERA registry point to a pathophysiological continuum [16]. Consequently, our present knowledge and definitions of this type of PH are largely fragmentary.
A similar situation exists for Group 3 (lung disease) and particularly for the mixed Group 5 PH patients. Patients with severe PH due to lung disease can be evaluated for individualised treatment in PH expert centres [1]; a retrospective analysis provided evidence of a survival benefit of targeted therapy [17]. In general, aside from patients in Groups 1 and 4, only highly selected patients should be treated off-label with PAH drugs in experienced centres, based on decisions made on a case-by-case basis. In isolated cases, improved symptoms, stabilised RV function and a favourable course are observed.
Table 2. Drugs used for treatment of pulmonary arterial hypertension (Group 1).
Future directions of PH therapy
Whereas established drug therapy of PAH addresses mainly vasoactive mechanisms, novel approaches target inflammation, proliferation, neurohormonal activation, and other pathways. Antiproliferative therapy with the tyrosine kinase inhibitor imatinib (known from therapy of chronic myeloid leukaemia and others) was shown to be effective in intensively pre-treated PAH patients, but did not gain approval because of adverse reactions [5]. A multitude of other agents (tacrolimus, rituximab, mineralocorticoid receptor antagonists, etc.) is currently under investigation, raising great expectations in terms of causal therapy of this disease that is by its nature proliferative and inflammatory [5]. A randomised study testing bisoprolol in patients with idiopathic PAH showed decreases in cardiac index and exercise capacity; thus, the results do not favour the use of ß-blockers in PAH patients [18].
PH due to heart failure with reduced ejection fraction is being addressed by a new multicentre trial of sildenafil in these patients [19]. Patients with PH due to left heart disease and elevated pulmonary vascular resistance are planned to be included, and the primary endpoint is improvement of exercise tolerance as measured by the established six-minute walk test. Improvement of haemodynamics and RV function are secondary objectives. In two previous trials, improved PAP was chosen as the primary endpoint but the results were inconclusive. Considering that PAP is not an established therapy goal in PH patients because an improved RV function can lead to a higher PAP and vice versa, the negative results of these studies were not surprising. Future studies should address RV function itself as the primary outcome measure, determined haemodynamically (cardiac output, right atrial pressure) or with imaging (for example, CMR), as this is what PH therapy should provide, i.e., improvement of RV function and thus symptoms and outcomes.
Supportive therapy of RV failure
Aside from therapy of the underlying disease such as PH or coronary artery disease, there is no known specific treatment for right heart failure. Volume management and decongestion are the predominant features of symptomatic treatment, requiring differentiated and consequent diuretic therapy as well as a close weight monitoring of the patient. Big differences exist among diuretic regimens, which should contain mineralocorticoid receptor antagonists, a thiazide diuretic and a loop diuretic in adequate dosage to reach euvolaemia. In advanced RV failure, cardiorenal syndrome caused by elevated central venous and intra-abdominal pressures commonly complicates volume excretion. Renal failure is characterised by the inability to ensure volume control in these patients, which may or may not be accompanied by azotaemia. If conservative management fails, renal support in the form of peritoneal dialysis should be considered. This process leads to an effective renal and abdominal decongestion, often resulting in improved renal function and regularly in a marked relief of symptoms. RV failure patients with a huge volume overload commonly present with tricuspid regurgitation (TR), frequently severe. In many cases, surgical repair of the tricuspid valve is prescribed, which misses the fact that TR is a kind of flag of volume overload. Consequent decongestion leads to reduction of TR severity in the majority of cases, whereas the surgical approach is risky and not indicated. In the few cases where primary TR causes RV failure, however, surgical repair may be the treatment of choice; the difficulty in discriminating one from the other is an unresolved clinical dilemma.
In the acute setting of RV failure, vasopressor and inotrope treatment may be required as well as mechanical circulatory support, for example in RV myocardial infarction or acute pulmonary embolism [20]. If PH as a cause of the acute RV failure is known, intravenous administration of sildenafil or epoprostenol may be an option for rapid reduction of RV afterload and circulatory stabilisation.
Conclusion
Despite substantial progress in treatment options for PAH and CTEPH, the effect of drug treatment on prevention of right heart failure and the beneficial effect on the prognosis of these patients remain limited. Early combination therapy with vasoactive agents is able to improve exercise capacity and quality of life and to delay manifestation of RV failure. To optimise therapy, close follow-up monitoring and consequent intensification of treatment are necessary. In every patient with precapillary PH, a thromboembolic origin of the disease should be excluded by ventilation-perfusion scintigraphy of the lungs, as the diagnosis of CTEPH offers the possibility of a cure by pulmonary endarterectomy. Even though a considerable proportion of surgical patients retain some degree of residual PH, most of them show a dramatic improvement that ranges up to “restitutio ad integrum”. In general, however, it is not currently possible to treat PAH causally, and this is one of the greatest present challenges of this disease.
Other forms of PH, in particular those due to left heart disease and to lung disease, are much more frequent than PAH and at the same time lack proven therapy options. Intensive research is underway in this field, as many patients are currently suffering from these diseases and doctors are dependent on experience-based attempts at treatment. In some cases, symptoms of RV failure can effectively be relieved and its progression can be slowed by targeted therapy. The majority of PH patients will eventually need supportive, symptomatic therapies for RV failure. RV function itself needs to be used as the primary outcome measure in future studies, as the goal of PH therapy should be improvement of right heart function and thus exercise capacity, quality of life and prognosis.
