















What is genetic testing for DCM?
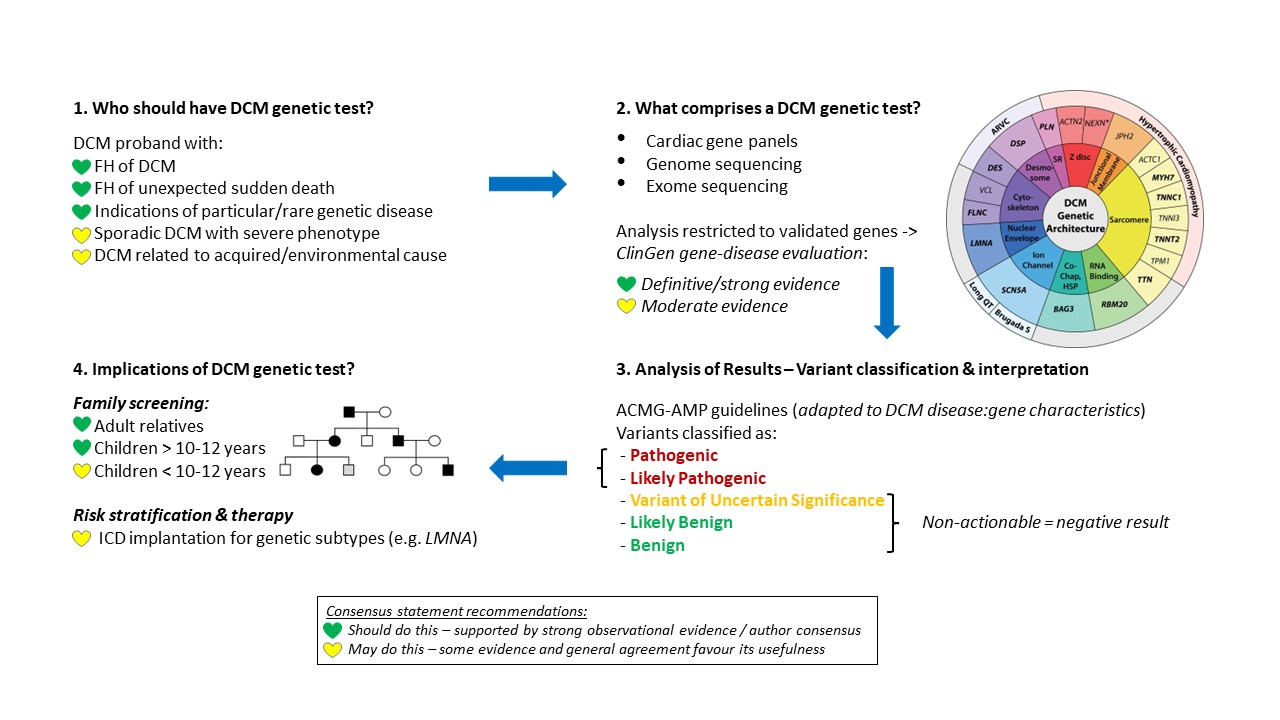
Genetic testing for DCM aims to identify the (usually one) rare pathogenic genetic variant that is the primary causal factor for disease in a patient. As per current guidelines, genetic testing is recommended for DCM probands with a family history of disease or sudden unexplained death or with clinical features suggestive of a particular/rare genetic disease. It may also be considered for patients with apparently sporadic DCM (especially at younger ages or with severe presentation) or those with DCM related to an acquired/environmental cause that may have genetic overlap (e.g. alcoholic cardiomyopathy).
How can genetic testing inform clinical management for patients and families?
If an unambiguous, disease-causing variant can be identified by genetic testing in a DCM patient, this can impact on clinical management in a number of ways:
- Confirming diagnosis: A positive genetic test result can confirm diagnosis of DCM in cases of phenotypic uncertainty or the presence of other environmental risk factors.
- Focused family cascade screening: Ongoing clinical screening of the patient’s family can be restricted to those with the variant and therefore at risk of developing DCM, enabling the half of relatives without the variant to be discharged and saving healthcare resources. As DCM is characterised by incomplete and age-dependent penetrance, clinical screening of variant carriers should be ongoing to detect early changes in phenotype and allow for optimal and early clinical management.
- Prognostic and therapeutic implications: Genetic testing results can influence clinical decision making for DCM patients. Pathogenic variants in genes like LMNA, FLNC, RBM20 and PLN have been associated with increased risk of ventricular arrhythmias and sudden cardiac death, and ICD implantation should therefore be considered in such cases.
- Reproductive planning: A genetic diagnosis can also be used to inform reproductive planning through prenatal or preimplantation genetic testing, particularly for variants associated with severe and phenotypes.
How is genetic testing performed for DCM patients?
For DCM genetic testing, the associated genes are usually sequenced as part of a gene panel (either specific to DCM/cardiomyopathies or a broader cardiac disease panel), although exome/genome sequencing are increasingly feasible and affordable options as primary sequencing assays. The latter options allow for re-analysis of a patient’s genomic data when new disease genes are identified and, in the case of genome sequencing, enable analysis of a broader spectrum of variant classes (e.g. to include intronic variants that may affect splicing).
While over one hundred genes have been implicated in DCM, consensus guidelines now recommend restricting clinical genetic testing to the 19 genes that are currently classified as having definitive, strong or moderate evidence for association with disease, based on the recently published ClinGen gene-disease re-evaluation process. These are the only genes in which actionable findings, i.e. pathogenic variants, can be routinely identified using stringent, clinical-grade variant classification guidelines.
How are the results of a genetic test interpreted?
Variants identified during clinical genetic testing are classified according to guidelines produced by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG-AMP). These guidelines provide a framework for how different lines of evidence (e.g. population frequency, functional assay validation, predictions of computational algorithms) should be used in evaluating the potential effect of a variant and the appropriate relative weighting for each. Depending on the nature and quantity of the evidence, variants will be classified as Benign (B), Likely Benign (LB), Uncertain Significance (VUS), Likely Pathogenic (LP) or Pathogenic (P).
There are a number of important aspects to note about the ACMG-AMP guidelines:
- The guidelines provide a broad framework for variant classification, but the specific application (and modifications where appropriate) needs to be defined for each disease (and disease-gene pair), e.g. which genes have a proven loss-of-function mechanism for DCM? An adaptation of the guidelines for DCM has been recently proposed (Morales et al, Circ Genom Precis Med 2020).
- As the application of the framework can differ between clinical laboratories, there can still be inconsistencies in variant classification – the ACMG-AMP guidelines at least provides the common “language” with which these differences can be evaluated.
- The guidelines are stringent in defining a variant as P/LP (i.e. clinically actionable), given the potentially adverse consequences of false positive results. This can make it difficult to reach an unambiguous classification for missense variants in particular, given the genetic heterogeneity of diseases like DCM and the relatively high rate of such variants in the general population. For DCM, the classification of missense variants in genes like MYH7, LMNA, DES and the troponin genes can be particularly challenging. More data sharing between labs and new computational and experimental techniques to improve variant classification are some of the ways to address this critical issue.
- Genetic testing and the interpretation of results is best undertaken in a multi-disciplinary setting that includes the expertise of cardiologists, clinical geneticists, genetic counsellors and genetic scientists.
Current limitations with genetic testing for DCM
Although genetic testing is now an established part of clinical management for DCM, its utility is currently limited by several factors:
- The yield of genetic testing, i.e. the proportion of patients where a causative and actionable variant is identified, is generally less than 30%. The remaining (majority) of cases are likely accounted for by (1) uninterpretable (i.e. VUS) or undetected variants in established disease genes, (2) as yet undiscovered disease genes or (3) non-Mendelian disease with a complex aetiology of genetic and non-genetic risk factors (this is likely to represent the majority of negative test cases).
- While the findings in DCM genetic testing do have some prognostic and therapeutic implications as described above, these are limited to a small number of genes / variant classes. More comprehensive and individualised risk stratification models (incorporating genetics, imaging and other clinical data) are required to fully harness the power of genetic profiling for DCM patients.
- Similarly, while a positive genetic test result can stratify family members for ongoing clinical screening, we are unable to predict which carriers of pathogenic variants are likely to develop disease, a particularly pressing issue for low penetrance genes like TTN and in the context of population screening where the likelihood of variant carriers becoming symptomatic will be even lower than within families.
- Current genetic testing is largely based on autosomal dominant modes of inheritance and the associated genes. An increasing number of genes related to recessive DCM are now being discovered (e.g. NRAP, JPH2) which will need to be incorporated into testing panels – these are likely to be particularly relevant for certain patient groups, like paediatric cases and patients from isolated populations or regions of high consanguinity.
