
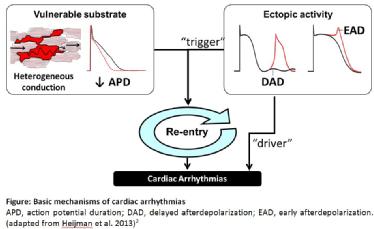
Ectopic activity is defined as abnormal impulse generation outside the sinus node. When ectopic activity occurs in a self-terminating manner, it may initiate arrhythmias by triggering reentry in a vulnerable substrate. Apart from the “trigger”, experimental and clinical evidence suggests that high frequency ectopic activity alone can maintain arrhythmias as a so called “driver”.(Ref. 5) Under these conditions, the surrounding tissue cannot keep up with the high frequency of the driver in a one-to-one manner and starts to fibrillate due to heterogeneous electrical impulse conduction (“fibrillatory conduction”). In several forms of AF radiofrequency ablation of the ectopic foci can successfully terminate the arrhythmia, thereby validating the importance of such “drivers” for the maintenance of these AF forms. (Ref. 5)
It is assumed that triggered activity is mainly caused by afterdepolarizations.(Ref. 3) They may occur during or after repolarization of the cardiac action potential (AP) and are termed early- and delayed afterdepolarizations, respectively. Early afterdepolarizations (EADs) occur when AP duration (APD) is largely prolonged, for example in patients with Long-QT-syndrome. Depolarizing cell-membrane Na+ or Ca2+-currents can recover from inactivation and allow Na+ or Ca2+ ions to move inward, causing membrane depolarizations. (Ref. 6)
Delayed afterdepolarizations (DADs) are caused by ‘spontaneous’ (non AP-triggered) Ca2+ release from the sarcoplasmic reticulum (SR) during diastole, generally due to SR Ca2+ overload or dysfunction of RyR2 (see newsletter from 31/07/2014). (Ref. 4, 7) The latter may be due genetic abnormalities in cardiac ryanodine receptors (RyR2) as in patients with catecholaminergic polymorphic ventricular tachycardia (CPVT) or due to RyR2 hyperphosphorylation as it has been shown for patients with heart failure and atrial fibrillation. Diastolic SR Ca2+ release events result in a transient inward current (Iti), largely carried by the Na+-Ca2+ exchanger (NCX1), which pumps in 3 Na+ for every Ca2+ extruded. The amplitude of the resulting DADs is determined by the magnitude of the Iti and the opposing repolarizing K+ currents responsible for maintaining the resting membrane potential. In cardiac tissue, the Iti-mediated depolarization also depends on electrotonic influences of neighboring cardiomyocytes and is larger when electrotonic coupling between myocytes is impaired. (Ref. 8) When a DAD reaches sufficient amplitude, reactivation of INa can occur, resulting in a triggered AP.
Reentry describes a continuous electrical impulse propagation around an electrically inactive tissue area. The healthy heart is protected from reentrant excitations by the relatively long refractory period, during which the tissue cannot be re-excited. In order to maintain reentrant excitations an appropriate arrhythmogenic substrate is necessary. The latter is characterized by a shortened refractory period, which is at the cellular level largely determined by the APD and the post-repolarization refractoriness controlled by the kinetics of the cardiac Na+-current. (Ref. 3, 4, 7) APD is determined by the delicate balance of depolarizing and repolarizing ion currents. Therefore, APD shortening can result from reduced activity of depolarizing inward currents (Na+- or Ca2+ currents, making the cell interior more positive) or from increased activity of repolarizing outward currents carried by K+ (making the cell interior more negative). Besides a shortened refractory period, slow electrical impulse propagation due to fibrosis or impaired electrical coupling between cardiomyocytes may also facilitate reentry. This provides more time allowing tissue to regain excitability. (Ref. 9)
Most of the currently available antiarrhythmic drugs inhibit cardiac ion-channels thereby reducing the likelihood of reentry occurrence. This may be achieved for example by prolonging the APD due to inhibition of repolarizing K+-currents (e.g. Amiodarone) or reduce excitability of cardiomyocytes by inhibiting Na+-currents (e.g. Flecainide). However, there is hope that targeting other arrhythmia mechanisms (e.g. SR Ca2+-leak) may lead to the development of safer and more effective antiarrhythmic therapeutic compounds. (Ref. 1, 2)