


The importance of early diagnosis of Fabry Disease
Early onset of FD signs and symptoms warrant prompt diagnosis, particularly because enzyme replacement therapy (ERT) is available. However, recognizing the early manifestations in clinical practice may be challenging for a variety of reasons. The disease presentation is generally heterogeneous, symptoms may resemble more common diseases, and major renal or cardiac dysfunction is uncommon in pediatric patients. Nowadays, diagnostic delay may still be considerable and patients often have to visit several medical specialists before a correct diagnosis is made. Recent data have shown that the overall diagnostic delay is about 15 years for both genders [5].
When to suspect the diagnosis of FD
- In children and adolescents: crisis of acute or chronic pain in the extremities (predominantly distal) which do not respond to analgesia; fever of unknown origin; heat, cold and exercise intolerance; chronic gastrointestinal disturbance; angiokeratomas; hypohidrosis; chronic pain or discomfort in the extremities; corneal whorls; retarded growth; microalbuminuria.
- In adults: persistence of the above symptoms; proteinuria; renal failure of unknown origin; unexplained LVH after 40 years; stroke; hearing loss [6].
When should the cardiologist suspect a diagnosis of FD cardiomiopathy?
Cardiac involvement is frequent in Fabry patients and is one of the most important causes of reduced life expectancy and disease-related death. Indeed, cardiac manifestations are reported in approximately 40-60% of patients with FD, and a prevalence of FD around 0.5-1.0% has been described in patients with hypertrophic cardiomyopathy (HCM) [7]. Therefore, the cardiologist should bear FD in mind when a diagnosis of HCM is made, whether the patient presents with the classic symptoms or only shows LVH, especially if data show typical echocardiographic or cardiac magnetic resonance imaging (MRI) findings.
All cardiac structures, including the myocardium, conduction system and valves, may be affected in patients with FD. Various cardiac manifestations have been reported, including symptoms of heart failure, angina pectoris (probably related to coronary microvascular dysfunction), and arrhythmias (the most common being atrial fibrillation and intermittent ventricular tachycardia). LVH is detected in ~50% of patients and has an earlier age of onset in males than in females. LVH is generally symmetrical, although asymmetric septal hypertrophy has been described. The typical pattern is a concentric thickening (with an end-diastolic wall thickness of up to 16 mm), without left ventricle (LV) outflow tract obstruction, but LV outflow tract obstruction has also been described. Other typical findings are prominent papillary muscles and preserved global ejection fraction combined with early stages of diastolic dysfunction. Right ventricular (RV) involvement is common in FD and ultimately progresses to severe diastolic RV dysfunction [7,8]. The end-stage cardiomyopathy is characterized by the coexistence of LVH, regional myocardial thinning and the presence of intramural fibrosis. In this case, characteristic changes seen on resting ECG are a positive Sokolow-Lyon index and a negative T-wave in the precordial leads (Figure 1a) [7].
The echocardiographic findings are not specific and also appear in patients with HCM of sarcomere origin and in other infiltrative cardiomyopathies and sometimes in the context of long-standing hypertensive heart disease. Pieroni et al [9] described the echocardiographic “binary appearance” of left ventricular endocardial border, more frequent in the interventricular septum, representing a sensitive and specific diagnostic hallmark of FD cardiomyopathy. However, a recent study in a large cohort of patients with FD has shown low sensitivity of this echocardiographic sign for diagnosis and it cannot adequately distinguish between patients with FD and patients with other causes of LVH (Figure 1b) [10].
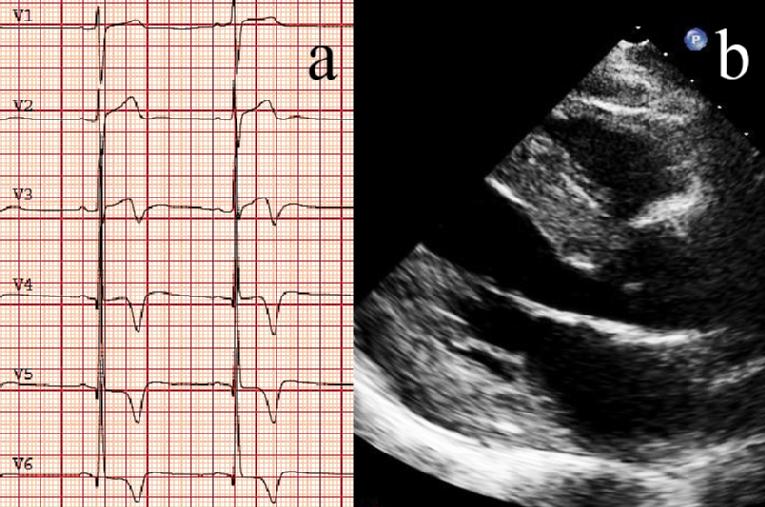
Figure 1. Typical electrocardiography (a) and echocardiography (b) images of a patient with Fabry cardiomyopathy. a) Positive Sokolow-Lyon index as well as the T-wave inversion in V3-V6. b) Long-axis view showing severe left ventricular hypertrophy.
Cardiac MRI is especially important to screen for myocardial replacement fibrosis by gadolinium late enhancement (LE) imaging. A higher relative concentration of gadolinium is found in myocardial areas with fibrosis compared to unaffected myocardium. In general, in both genders, pathological LE is mostly limited to posterior and lateral segments, with different distensions towards the apex but not reaching the apical segment. The assessment of myocardial fibrosis may help us to the diagnosis, is essential for staging the cardiomyopathy and is necessary for monitoring therapy effects. This is particularly important for female patients, who often develop myocardial fibrosis despite otherwise non-hypertrophic myocardium [7].
If clinical examination raises a suspicion of FD, diagnosis is confirmed using a combination of biochemical and molecular testing [2,3].
How to confirm the diagnosis
α-Gal A deficiency should preferably be measured in leucocytes. While this can be reliably established in other enzyme sources such as dried blood spots and plasma, leucocytes are superior in estimating residual α-Gal A activity. a-Gal A enzyme activity dosage on dried blood spots using a filter paper test has been proposed as an alternative diagnostic test and has been found to be an accurate assay. The accuracy of this test, using the threshold of enzyme activity <40% of controls, reaches a sensitivity and specificity of 100% in men, whereas in women sensitivity was lower (66%). Moreover, samples are easy to transport and are stable at room temperature for at least 20 days, making them suitable for screening patients at risk of FD, and could also be used as a source of DNA. Normal enzyme values differ depending on the enzyme source, substrate concentrations, and assay variables. However, the diagnosis of FD in women can occasionally be made using these methods in cases in which a markedly decreased residual enzyme activity is evidenced, but the genetic analysis of the gene is required, because females can have normal to very low a-Gal A activity [11].
Although in the past low α-Gal A activity has been considered sufficient for diagnosis in males, the presence of a common pseudodeficiency allele, D313Y, which results in low α-Gal A plasma activity, and slightly reduced leukocyte enzyme activity suggests that a diagnosis of FD disease should not be finalized until a disease-causing GLA mutation is identified [12]. Once a GLA gene mutation has been identified in a proband, targeted mutation analysis can be used to diagnosis at-risk male and female family members for that specific family mutation. While the pathogenicity of some GLA mutations is well described, the subjects identified through screening often have a GLA genetic variant/mutation of unknown significance (GVUS). Interestingly, most males with such GVUS demonstrate significant residual a-Gal A enzyme activity, in contrast to the absent or near absent enzyme activity in classically affected males [2].
If clinical suspicion is high with normal enzyme activity or without diagnostic mutation, the finding of elevated values of plasma/urine Gb3 or deposits in a target organ (such as kidney biopsy, skin biopsy, etc.) could be helpful [6]. European consensus recommendations on diagnosis in adults with LVH and GVUS, which include the diagnostic criteria for a definite diagnosis of FD, have recently been published [2,3]. After a diagnosis of FD has been confirmed, clinical evaluation should involve a multidisciplinary team of subspecialists, which should be coordinated by a physician experienced in the care of patients with FD [6].
The role of the cardiologist in the screening of FD: early damage indicators
The cardiologist may face different situations (Figure 2):
- Patients with a diagnosis of FD referred to rule out cardiac involvement.
- Patients diagnosed with HCM.
- Diagnosis of FD via analysis of family history.
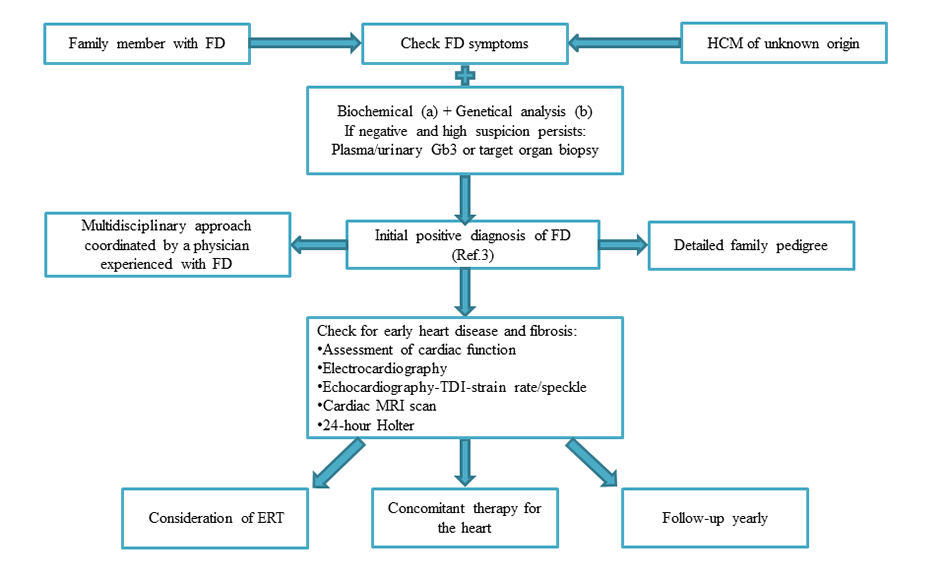
Figure 2. Algorithm for the diagnosis and assessment of patients with Fabry disease focusing on the role of the cardiologist (see text for more detail). The initial point of presentation will vary. Leucocyte/Plasma alpha-galactosidase A activity or enzyme activity dosage on dried blood spots using filter paper test b. Analysis of the GALA gene or specific familiar mutation if previously detected. FD, Fabry Disease; HCM, hypertrophic cardiomyopathy; Gb3, globotriaosylceramide; TDI, Tissue Doppler Imaging; MRI, magnetic resonance imaging
Patients with a diagnosis of FD referred to rule out cardiac involvement
As we have described above, 2D echocardiography and cardiac MRI are diagnostic tools which help us to detect LVH, the evaluation of left atrium and valves, diastolic dysfunction in the case of echocardiography, as well as fibrosis in the case of MRI. These findings allow the diagnosis once there is a manifest cardiac involvement, but do not detect incipient cardiac involvement with Gb3 storage at this level for years. That is why, in the last decade, new techniques have been developed to assess indirect signs of cardiac involvement before LVH or fibrosis appear.
Tissue Doppler imaging (TDI)
Several studies have identified reduced TDI velocities in Fabry patients without echocardiographic evidence of Fabry-related cardiomyopathy. The observation that TDI velocities are further reduced in Fabry patients with LVH has suggested that TDI abnormalities might be an initial finding of cardiac involvement in FD. TDI has been reported to distinguish between patients with FD with and without LVH and relatives without the FD mutation. It has been reported that TDI measurements can distinguish between FD patients without LVH and healthy subjects without the disease [13,14].
Strain-rate imaging and 2D speckle tracking
These two echocardiographic techniques can indirectly assess replacement fibrosis but do not allow an exact quantification of the amount of fibrosis. However, it has been shown that a systolic longitudinal strain value of more than 16.5% in the typical posterolateral region makes replacement fibrosis extreme unlikely, whereas a value lower than 12.5% is a related strong indicator of replacement fibrosis [7] .
Biomarkers
The most important biomarker is lyso-Gb3, a degradation product of the accumulated Gb3. It has been shown that plasma lyso-Gb3 concentration is severely elevated in all classic Fabry patients. In addition, it is slightly elevated in atypical late onset mutations. Overall, the level of lyso-Gb3 is considered to predict the potential severity of unknown mutations. This is particularly helpful in detecting atypical late onset forms and women with a-GalA activity around the normal threshold. Additionally, lyso-Gb3 is viewed as an independent risk indicator for LVH in female patients [7].
Patients diagnosed with HCM
Among patients diagnosed with HCM, cardiac variants of FD are not exceptional and might benefit from specific ERT. According to the latest 2014 European Society of Cardiology Guidelines on Diagnosis and Management of Hypertrophic Cardiomyopathy, the most common metabolic disorder in adults with HCM is FD [15,16]. In one study using a systematic genetic screening in 90 HCM probands, 59 without sarcomere gene mutations, a-Gal A mutations were found in three out of 90 (3%) HCM families and in two of 20 (10%) women without sarcomere gene mutations; none of the probands presented other indices of FD. This finding supports systematic testing for FD among patients diagnosed with HCM [17].
Diagnosis of FD via analysis of family history
Increasingly, patients are also being diagnosed via pedigree analysis after diagnosis of another family member. The pedigree review from the lysosomal storage disease centers and contributing family members found that, on average, there are at least five family members who are diagnosed with FD following the diagnosis of a proband. A medical pathway beginning with the identification of at-risk family members through a detailed family pedigree can lead to earlier and more effective treatment of FD. In order to identify affected family members and obtain the benefits of early diagnosis, there are several steps which must be taken. First, a detailed family history focused on inheritance patterns of the X-linked condition and Fabry-related symptoms of family members should be produced in order to identify at-risk family members. Next, the proband should assist with contacting at-risk family members and their physicians to offer detailed information on testing for FD. Targeted mutation analysis can be used to diagnosis at-risk male and female family members for that specific family mutation [18].
Finally, after confirmatory testing, the physicians and the patient should work together to develop a lifetime monitoring and treatment schedule that follows the expert recommendations produced for the treatment of FD to fulfill the health benefit resulting from early diagnosis [4].
How should disease progression be followed?
All patients with FD should be followed regularly, regardless of disease status or treatment protocols. The course of the disease varies, even among family members with the same mutation. Although signs and symptoms will dictate the frequency and extent of follow-up, lack of overt symptoms does not preclude the need for an annual comprehensive medical evaluation because kidney, cardiac, or cerebrovascular function can decline so insidiously. The cardiac tests which should be carried out annually are an electrocardiogram, an echocardiogram with DTI and a cardiac MRI scan. Since many Fabry patients develop bradycardias, a 24-hours Holter ECG monitoring should be conducted before and during therapy, especially if symptoms like dizziness or syncope are reported. A cardiac stress test can also be performed [6].
Conclusions
Significant morbidity and mortality are associated with FD, and Fabry cardiomyopathy is an important organ manifestation of the disease. Cardiac diagnosis, focused on myocardial fibrosis, is frequently difficult and a comprehensive diagnostic approach is necessary. Given the potential benefits of early treatment, early diagnosis and treatment to modify the natural history of the disease is desirable.