

Background
Screening is possible in well-defined patient groups at high risk of developing PAH. These include patients with connective tissue diseases (CTD), congenital heart disease, chronic liver disease, and HIV infection. Prognosis of PAH is usually quite poor, with an estimated survival of 70% and 50% at 3 and 5 years of diagnosis. When diagnosed, the vast majority of patients suffering from PAH are in WHO FC III and IV (confortable at rest, or mostly bedbound), which predict poorer survival.
I - DIAGNOSTIC ALGORITHM
A) Suspicion and Exclusion:
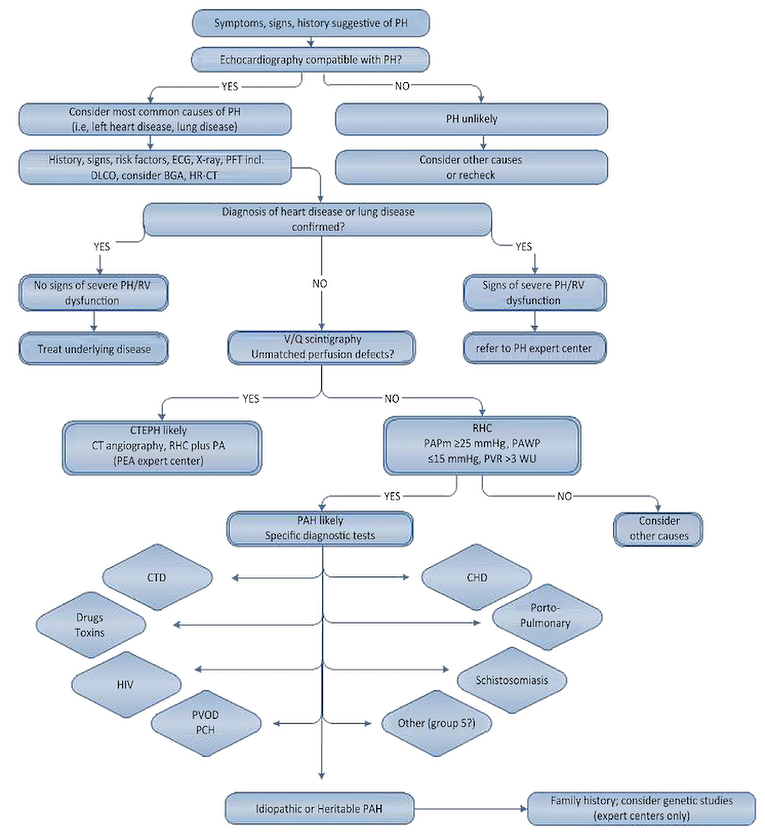
Here presented is the latest world’s symposium on pulmonary hypertension’s working group diagnostic algorithm proposal (Figure 1) (1).
First, there is clinical suspicion.
Then, the transthoracic echocardiogram will lead us to a diagnosis of pulmonary hypertension (PH). Probable PH in the echo must then lead the way to a diagnosis of exclusion, ruling out:
- Heart (group 2) or lung disease (group 3), which are the most common causes of PH.
- We must also to rule out LV diastolic dysfunction, sleep apnea and COPD (2).
- Chronic thromboembolic pulmonary hypertension (CTEPH; group 4) must be ruled out (or confirmed). If CTEPH is ruled out, diagnosis of pulmonary arterial hypertension (PAH, group 1) is very probable and must be confirmed with right heart catherisation. Then we will identify which type of PAH we are presented with.
B) Addressing pulmonary pressures:
Before performing RHC, every entity that may increase pulmonary pressures should be ruled out or treated. Daily practice examples include every situation that determine left ventricle (LV) diastolic dysfunction (high blood pressure, overweight, high heart rate if atrial fibrillation is present…), and respiratory disorders (sleep apnea, COPD…). Primary disease must be treated, and PAH specific therapy must be avoided. The increment in pulmonary pressures secondary to those entities will decrease after correct management.
C) Establishing the diagnosis:
Transthoracic echocardiography is still the most important non-invasive screening tool to assess the possibility of PAH, but right heart catheterisation (RHC) is still mandatory to establish the diagnosis (3).
Limitations and prevalence: Our current diagnostic tools are often insufficient to provide a clear distinction between PAH and PH due to left heart disease. Fluid overload during RHC helps to identify those patients with PH due to LV diastolic dysfunction.
The estimated prevalence of PAH is 16 cases per million adult inhabitants (MAI) and CTEPH is 3.2 cases, whereas Group 2 (heart) and 3 (lung disease) PH are very common and concern over more than 90% of PH detected in echocardiogram lab. The treatment goal in these patients will be adequate management of the underlying disease. (4)
Figure 1. Diagnostic algorithm of pulmonary arterial hypertension.
II – Therapy
General measures:
Added to the measures previously described here, the following should be avoided:
- Pregnancy should be avoided because it remains associated with a substantial increase in mortality and
- Excessive physical activity that leads to distressing symptoms. However rehabilitation and exercise training are openly recommended (Class I with a Level of Evidence). Supportive therapy and treatment with channel blockers are described in previous article.
Pharmacology: Drugs are classified according to the grade of recommendation and the level of evidence on the basis of published randomised clinical trials (RCTs). They are also stratified according to WHO-FC. As head-to-head comparisons among different compounds are not available, no evidence-based first line treatment can be proposed for either WHO-FC II or III patients. The choice of the drug may depend on: the approval status, the labeling, the route of administration, the side effect profile, patient’s preferences, physician experience, and cost.
Vasoreactive patients: Therapy with PAH-approved drugs must be initiated in PAH patients who are not vasoreactive or are vasoreactive but not responding appropriately to cardiac channel blockers (CCBs).
Limitations: The treatment algorithm does not apply to patients in other clinical groups (i.e. groups 2-5), and in particular not to patients with PH associated with left heart disease (group 2) or with lung diseases (group 3). In these groups, specific pulmonary vasodilator therapy could be deleterious. Again, the accurate diagnosis is mandatory and LV diastolic dysfunction and/or pulmonary disease must be ruled out before indicating specific therapy.
III - Addressing the clinical response to initial therapy
Evaluating initial response: After initial therapy, the clinical response must be reassessed at 3 to 6 months. Response is based on the evaluation of various parameters: WHO-FC, exercise capacity, cardiac index, right atrial pressure, NT-proBNP plasma levels, echocardiographic parameters, and perceived need for additional/change of therapy.
Patient evolution: If the patient’s state does not improve or deteriorates, combination therapy (using 2 or more classes of drugs simultaneously) should be considered. Three separate signaling pathways are known to be involved in the disease: the prostacyclin pathway, the endothelin pathway, and the NO pathway. In WHO-FC IV patients, initial combination therapy may also be considered.
Sequential therapy: Combinations of approved drugs and additional interventional procedures, in case of inadequate results, must be considered. Combination therapy can either include an ERA plus a PDE-5i or a prostanoid plus an ERA or a prostanoid plus a PDE-5i. The patterns to apply combination therapy may be sequential or initial (upfront). Sequential combination therapy is the most widely used strategy. From monotherapy there is addition of a second and then third drug in cases of inadequate clinical results or in cases of deterioration. Sequential combination therapy has been allotted a grade of recommendation I and level of evidence A in PAH patients with inadequate clinical response to initial monotherapy. In case of inadequate clinical response, sequential combination therapy should be considered.
Epoprostenol: Continuous iv epoprostenol is recommended as first line therapy for WHO-FC IV PAH patients because of the survival benefit in this subset.
Goal-oriented therapy: A structured prospective program to evaluate the adequacy of clinical results is the so-called “goal-oriented therapy”, a treatment strategy that uses known prognostic indicators as treatment targets (6). The therapy is considered adequate only if the targets are met. The key difference between goal-oriented therapy and non-structured approaches is that patients who are stabilised, or even those who improve slightly, can still receive additional therapy if treatment goals are not met. The goal-oriented treatment strategy is shown in Table 1. Better prognosis in patients achieving these goals has been confirmed.
Table 1 – Variables and criteria used to determine response to therapy and prognosis in patients with PAH
| Variable | Criteria |
|---|---|
|
Functional class |
I or II |
|
Echography/CMR |
Normal/near-normal RV size and function |
|
Hemodynamics |
Normalisation of RV function (RAP <8mmHg and CI>2.5 to 3.01/min/m2 |
|
6 min walk distance |
>380 to 440 m; may not be aggressive enough in young individuals |
|
Cardiopulmonary exercise testing |
Peak VO2>15ml/min/kg and EqCO2< 45l/min/I/min |
|
B-type natriuretic peptide level |
Normal |
Legend: CI=cardiac index; CMR=cardiac magnetic resonance; EqCO2=ventilator equivalent for carbon dioxide; PAH=pulmonary arterial hypertension; RAP=right atrial pressure; RV=right ventricular; VO2=peak oxygen consumption.
Lung transplantation should be considered for those patients who fail on maximal medical therapy and remain in WHO-FC III or IV. Both heart-lung and double-lung transplantation have been performed for PAH. Currently the vast majority of patients worldwide receive bilateral lungs. Eligibility for lung transplantation should be considered after an inadequate clinical response to the initial monotherapy. Referring the patient for lung transplantation soon after the inadequate clinical response is confirmed on maximal combination therapy should be the next step.
Ballon atrial septostomy (BAS). The creation of an interatrial right-to-left shunt can decompress the right heart chambers, and increase LV pre-load and cardiac output. In addition, this improves systemic O2 transport despite arterial O2 desaturation and decreases sympathetic hyperactivity. It benefits patients in WHO-FC IV with right heart failure refractory to medical therapy or with severe syncopal symptoms. It may also be considered in patients awaiting transplantation or when medical therapy is not available. BAS should be regarded as a palliative or bridging procedure to be performed only by centers with experience.
Acute vasoreactivity testing should be performed in all patients with PAH (group 1), although patients with idiopathic PAH, heritable PAH, and PAH associated with anorexigen use are the most likely to exhibit an acute positive response. Vasoreactive patients should be treated with high and optimally tolerated doses of CCBs; adequate response should be confirmed after 3 to 4 months of treatment. Another RHC is needed to confirm improvement in hemodynamic parameters added to clinical, functional and structural data. Non-responders to acute vasoreactivity testing who are in WHO-FC II should be treated with an oral compound. Non-responders to acute vasoreactivity testing, or responders who remain in (or progress to) WHO-FC III, should be considered candidates for treatment with any of the approved PAH drugs.
conclusions
Pulmonary arterial hypertension (PAH) is rare yet severe and must be suspected in any patient with unexplained dyspnea. Physician awareness of the diagnostic algorithm we propose will provide for accurate diagnosis and early targeted therapies started before the development of significant right heart failure – thus allowing for no delay between onset of symptoms and diagnosis.