
Torsades de pointes (TdP) is a potentially life threatening polymorphic ventricular tachycardia, electrocardiographically characterised by QRS axis undulations over runs of several beats, with a specific twist of the QRS complex around the isoelectric baseline occurring on an abnormal QT-prolongation (usually to 600 msec or greater). The arrhythmia usually terminates spontaneously, with the exception of rare cases of degeneration into ventricular fibrillation. The episodes are rate-dependent (1) and often repetitive.
Several reports have associated TdP and bradycardia, with atrio-ventricular block especially (2, 3) - 5% to 30% of patients with atrio-ventricular block presenting TdP (4-6). The feature common to atrio-ventricular block and TdP seems to be the presence of QT interval prolongation (7). This particular category of patients, presenting atrio-ventricular block induced – QT prolongation, has the most elevated risk of developing TdP. Moreover, in patients with atrio-ventricular block, QT interval prolongation was identified as an independent predictor of symptoms, including cardiac arrest and syncope (8).
Fig. 1. Holter recording in a patient with post-operative atrio-ventricular block and typical TdP.
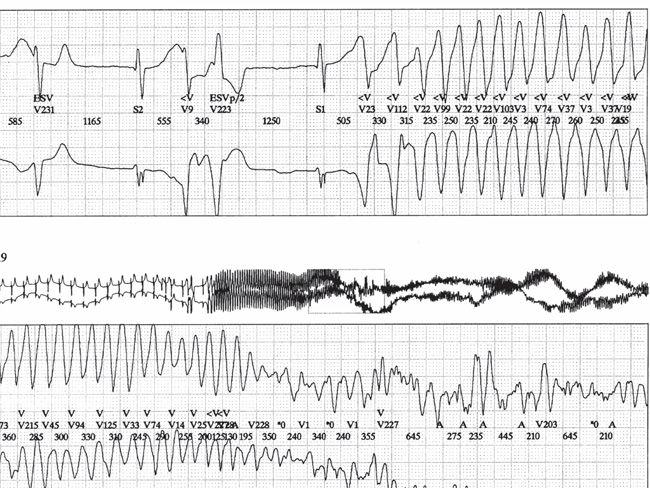
Note the association of long QT interval, ventricular bigeminism, bradycardia, and bradycardia-dependant polymorphic ventricular tachycardia degenerating into ventricular fibrillation.
1) Risk factors for atrio-ventricular block-induced torsades de pointes
The most commonly encountered risk factors for TdP are: female gender, advanced age, bradycardia as well as a number of metabolic disorders and therapeutic agents (9).
The gender-specific preponderance of females developing drug-induced torsades de pointes when treated with antiarrhythmic drugs or during spontaneous bradyarrhythmias is well documented. Usually, women have a longer corrected QT interval than men.
Several endocrine disorders as well as a number of electrolytic imbalances have been incriminated as TdP facilitators (hypothyroidism, mental anorexia, hypokalaemia, hypomagnesaemia, hypocalcaemia and acidosis).
A great number of drugs are currently considered “torsadogenic”. Almost all of these drugs have an IKr channel blocking effect, which explains QT interval prolongation. These agents belong to both “cardiotropic” and “non-cardiotropic” drugs (an exhaustive list is available here).
2) Genetic bases of atrio-ventricular block-induced torsades de pointes
Only a minority of patients with atrio-ventricular block develop TdP and these patients have longer QT intervals. These observations suggest a genetic predisposition of the affected subjects.
Common clinical features and the presence of long QT intervals in patients with congenital long QT syndrome and in those with atrio-ventricular block–induced TdP suggest that patients with atrio-ventricular block-mediated QT-related arrhythmia could have latent congenital long QT syndrome or a vulnerable genetic polymorphism (10).
Several similarities have been found in experimental models of atrioventricular block–induced TdP and specific forms of human congenital long QT syndrome: enhanced susceptibility to acquired TdP, adrenergic dependence of the arrhythmia, typical T-wave patterns during prolonged QT intervals and reduction of IKs current in dogs with chronic atrio-ventricular block closely resemble the clinical characteristics of the LQT1 or LQT5 forms of human congenital long QT syndrome. Frequent episodes of spontaneous TdP, abnormal QTU complex, and reduction of both IKr and IKs in a rabbit model of atrio-ventricular block closely resemble the clinical characteristics of the malignant form of human congenital long QT syndrome caused by double mutation of HERG and KCNQ1 (11).
Bradycardia-related repolarisation abnormalities appear to be allelic variants in the coding region of congenital long QT syndrome genes, variants that can be identified in 10–15% of affected subjects. A recent retrospective study showed that 14% to 18% of subjects with atrio-ventricular block-related QT-interval prolongation were carriers of a genetic mutation in genes coding for potassium channels. Another study reported genetic mutations involving HERG and SCN5A in 36% of patients with complete atrioventricular block and TdP (12). In fact, the most commonly encountered mutation in this setting is in HERG genes, coding for potassium channel IKr.
3) Pathophysiology of atrio-ventricular block-induced torsades de pointes
Although a slow ventricular rate may be a major factor for inducing abnormal QT prolongation, the mechanisms of QT prolongation in patients with bradycardia-related TdP are poorly understood.
Clinical and experimental data show that the duration of atrio-ventricular block is an important determinant of susceptibility to acquired TdP (13, 14). Concomitant to the duration of atrio-ventricular block, the likelihood of structural and electrical alterations in myocardial cells may increase.
The cardiac remodelling processes induced by chronic slow heart rates offer both the “trigger” and the “substrate” for the occurrence of TdP. Bradycardia-induced volume overload causes a biventricular eccentric hypertrophy (15) and consequently a non-homogenous lengthening of the ventricular action potential duration. While electrical and structural changes in chronic atrio-ventricular block are aimed at maintaining cardiac function, they also predispose to QT prolongation. These adaptations, alone or synergistically, increase the risk of early after-depolarisations and / or delayed after-depolarisation and therefore the risk of TdP.
Much information has been offered by a rabbit model of atrio-ventricular block induced by transcatheter radiofrequency ablation of atrio-ventricular node (11). Chronic endocardial ventricular pacing at near-physiologic rate was installed immediately after the ablation procedure. Identical amplitudes of all repolarising currents were found in both atrio-ventricular block group and sham-operated group.
Moreover, a similar down-regulation in the affected currents regardless of ventricular cavity stimulation (left or right ventricle) was reported, and authors concluded that secondary electrical remodelling was unrelated to the loss of atrio-ventricular synchrony, bradycardia alone sufficing to turn on the electrical remodelling process.
Conclusion:
Investigations of the clinical aspects and molecular mechanisms of long QT syndrome have provided novel and important insights into the basis of ventricular arrhythmias and shown how small perturbations in ion flow can have important consequences in human health. Common clinical features and the presence of long QT intervals in patients with congenital long QT syndrome and in those with atrio-ventricular block–induced TdP suggest that patients with atrio-ventricular block-mediated QT-related arrhythmia could have latent congenital long QT syndrome or a vulnerable genetic polymorphism. We recommend systematically genotyping patients with acquired long QT syndrome and TdP. Known risk factors for TdP, such as metabolic disorders or “torsadogenic” drugs should be rigorously avoided. Ventricular pacing at age-appropriate rates may be used to normalise QT intervals and prevent TdP.