Summary of the paper
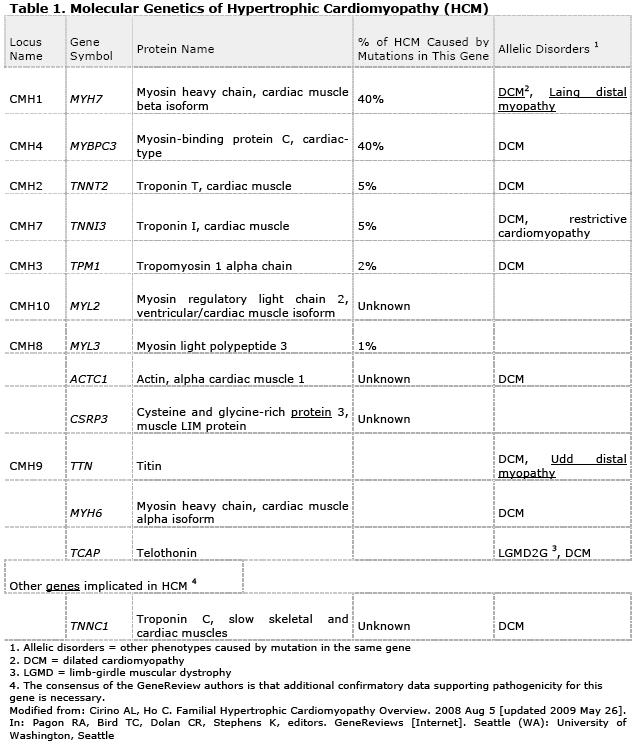
Girolami et al. (6) studied a total of 488 unrelated index HCM patients who underwent screening for myofilament gene mutations by direct deoxyribonucleic acid sequencing of 8 genes, including myosin binding protein C (MYBPC3), beta-myosin heavy chain (MYH7), regulatory and essential light chains (MYL2, MYL3), troponin-T (TNNT2), troponin-I (TNNI3), alphatropomyosin (TPM1), and actin (ACTC).
Of the 488 index patients, 4 (0.8%) harbored triple mutations, as follows: MYH7-R869H, MYBPC3-E258K and TNNI3-A86fs in a 32-year-old woman; MYH7-R723C, MYH7-E1455X and MYBPC3-E165D in a 46-year old man; MYH7-R869H, MYBPC3-K1065fs and MYBPC3-P371R in a 45-year old woman; and MYH7-R1079Q, MYBPC3- Q969X and MYBPC3-R668H in a 50-year old woman.
One patient had a history of resuscitated cardiac arrest, and 3 had significant risk factors for sudden cardiac death, prompting the insertion of an implantable cardioverter-defibrillator in all 4, with appropriate shocks in 2 patients. Moreover, 3 of the 4 patients presented a severe phenotype with progression to end-stage HCM by the fourth decade requiring cardiac transplantation (n = 1) or biventricular pacing (n = 2). The fourth patient, however, presented a clinically mild disease.
The authors concluded that HCM caused by triple sarcomere gene mutations was rare but conferred a remarkably increased risk of end-stage progression and ventricular arrhythmias, supporting an association between multiple sarcomere defects and adverse outcome. Comprehensive genetic testing might provide important insights to risk stratification and potentially indicate the need for differential surveillance strategies based on genotype.
Comment
These results strengthen previous observations and are a stimulus for further research in this direction. Multiple mutations have been found to occur in either the same gene (compound heterozygotes) or in different genes (double heterozygotes) in up to 5% of families with familial HCM. Importantly, individuals who carry 2 disease-causing familial HCM mutations have a more severe disease, including earlier onset, more severe left ventricular hypertrophy and heart failure and a higher rate of sudden death events (resuscitated cardiac arrest or sudden death). These preliminary observations suggest that the number of genes identified in an individual patient with familial HCM may be an important determinant of phenotype severity and clinical outcome of the disease. Tsoutsman et al. (7) developed, characterized and investigated the pathogenic mechanisms of double mutations in a double-mutant murine model of familial HCM. The mortality rate in TnI-203/MHC-403 mice was 100% by age 21 days. At age 14 days, TnI-203/MHC-403 mice developed a significantly increased of heart weight to body weight ratio, marked interstitial myocardial fibrosis, and increased expression of atrial natriuretic factor and brain natriuretic peptide compared to nontransgenic, TnI-203 and MHC-403 littermates. By age 16 to 18 days, TnI-203/MHC-403 mice rapidly developed a severe dilated cardiomyopathy and heart failure, with inducibility of ventricular arrhythmias, which led to death by the age of 21 days. The authors concluded that TnI-203/MHC-403 double-mutant mice developed a severe cardiac phenotype characterized by heart failure and early death.
It is noteworthy that 3 of the 4 patients (75%) with triple mutations in Girolami’s series developed an end-stage phenotype with severe LV dysfunction. These 3 patients had originally presented with severe LV hypertrophy by the age of 25. In the next 10 to 20 years however, they developed marked cardiac remodelling characterized by restrictive physiology, atrial dilation, and systolic dysfunction associated with progressive LV wall thinning and fibrosis; 1 patient required cardiac transplantation. By comparison, only 29 of 488 patients or 6% of the overall genotyped population with HCM seen at the same centre developed end-stage disease, with a similar prevalence to that in other reports. This suggests that triple mutations confer a 14-fold increase in risk of developing end-stage disease. Furthermore, all 4 probands with triple mutations had significant ventricular arrhythmias, prompting the implantation of a defibrillator for primary or secondary prevention of sudden cardiac death. Notably, 1 patient had been resuscitated from cardiac arrest, and another received multiple appropriate interventions after ICD implantation. Overall, the clinical course of HCM patients with triple mutations strongly supports the concept that multiple sarcomere defects might be associated with a more severe clinical phenotype and disease course.
A plausible explanation of the adverse consequences of complex genotypes is that multiple abnormal myofilament proteins might result in more profound derangement of sarcomere mechanics, myocardial energetics, and cardiomyocyte dysfunction. In addition, as the authors remark, to other pathophysiological mechanisms that might intervene, such as greater impairment of microvascular function due to adverse remodeling of the coronary arterioles, leading to recurrent myocardial ischemia and replacement fibrosis. Two of the patients reported with triple mutations had evidence of severe microvascular dysfunction and blunted myocardial perfusion preceding the development of LV wall thinning and systolic dysfunction, which is consistent with this hypothesis.
The TNNI3-203/MHC-403 double-mutant mouse model recently reported by Tsoutsman et al. (7), buttresses the concept of a gene dosage effect resulting in more severe clinical phenotypes. In this model, although each mutation by itself was linked to a hypertrophic phenotype, the presence of both mutations rapidly led to LV dilation, severe heart failure, and premature death. Additionally, downregulation of mRNA levels of key regulators of Ca² homeostasis in TnI-203/MHC-403 mice was observed. Increased levels of phosphorylated STAT3 were observed in TnI-203/MHC-403 mice and corresponded with the onset of disease, which suggests a possible cardioprotective response.
Mutations in sarcomeric genes lead to activation of intracellular signaling mechanisms, cardiac remodeling, and changes in contractile function. Signaling through the latent transcription factor signal transducer and activator of transcription (STAT) 3 has been implicated in linking cardiac myocyte remodeling with various extrinsic and intrinsic stimuli. Specifically, STAT3 activation, characterized by phosphorylation on a specific tyrosine residue (Y705), promotes cardiac myocyte hypertrophy both in cell culture systems and in animal models. Previous studies in murine models have indicated a role for STAT3 in cardiac protection, specifically with transgenic cardiac-specific STAT3 over-expression transducing a protective signal against cardiomyopathy after treatment with the antitumor drug doxorubicin in vivo and enhancing vascular formation in the heart in vivo to mediate additional cardiac adaptation under conditions of stress. Consistent with this role of STAT3 in cardiac protection, the cardiac-specific knockout of STAT3 enhanced susceptibility to cardiac injury after myocardial ischemia, with reduced cardiac function and increased mortality and resulted in higher sensitivity to inflammation, cardiac fibrosis, and heart failure with advanced age. In human hearts with end-stage dilated cardiomyopathy, altered STAT3 levels and activation have also been observed. Taken together, the results of these studies suggest that STAT3 may be an important mediator in the development of cardiac hypertrophy and, through its modulation of cardiac cell death, the subsequent progression to heart failure.
The pedigrees reported in the study by Girolami et al. highlight the complexity inherent to the HCM disease process and challenge conventional wisdom regarding the real clinical impact of single sarcomere gene mutations. For example, although most of their patients with triple mutations exhibited a severe phenotype and progressive disease, Patient #4 had modest LV hypertrophy and mild symptoms at age 50 years. Furthermore, several relatives of the index patients had an adult expression of the disease, despite carrying double mutations themselves. Such a discrepancy suggests that certain DNA variants might not be capable of causing disease in isolation but potentially exert modifying effects on disease expression, in combination with other mutations. The demonstration of such a hypothesis is hindered by the objective difficulty, inherent to all genetic studies in HCM, of proving which of the identified sequence variants are truly pathogenic and to what extent. However, the striking phenotypic expression and markedly increased prevalence of end-stage remodelling observed in this cohort study indicate that the multiplicity of variants importantly contributed to disease pathogenesis and clinical outcome.
Conclusion:
On the basis of the present findings, a comprehensive sarcomere mutational screening might provide important clues for risk stratification and potentially indicate the need for differential surveillance strategies based on genotype. Screening should probably not be stopped after the identification of one mutation, especially in families with a particularly severe phenotype, but should be continued on the same gene and at least on the 2 major genes. Additionally, due to the fact that the age at onset, the degree of hypertrophy, and the prognosis was related to the number of mutations in the families reported, before establishing phenotype-genotype correlations it seems necessary to check for a complex genetic status to better understand the broad expressivity of the disease and give these families better genetic counselling.