
Cardiac regeneration in fish and amphibians
Urodeles, such as salamanders and newts, are well known for their capacity to regrow their tails after loss during escape from predators. The regenerative capacity of salamanders goes far beyond this, and some salamander species have been shown to regenerate their limbs, jaws, spinal cord and hearts (Brockes and Kumar, 2005; McCusker and Gardiner, 2011; Tanaka and Reddien, 2011). Studies by Oberpriller and Oberpriller were among the first to describe the high regenerative potential of the salamander heart. After amputation of one eighth of the cardiac ventricle, signs of regeneration were detected within 30 days (Becker et al., 1974; Oberpriller and Oberpriller, 1974). In the initial response, a blood clot forms, followed by macrophage and lymphocyte infiltration and the deposition of extracellular matrix. Up to this point, the injury appears to follow the same cascade of events seen in the mammalian heart. However, at day 16 postinjury, recovery of the endocardium was observed at the site of injury and, most importantly, DNA synthesis was detected within CMs, indicating that the salamander heart is able to regenerate after injury. Similar findings were reported in another amphibian model, the frog (Rumyantsev, 1973).
These early observations were confirmed decades later in the zebrafish, which was then emerging as an important model organism in basic research. With its small size, the large number of embryos per clutch, and the ease of studying the development of its transparent embryos, the zebrafish offers the possibility to perform large-scale genetic and pharmacological screens in a vertebrate animal model and has therefore become an established model organism in cardiovascular research (Bakkers, 2011). The adult zebrafish heart can regenerate completely after resection of up to 20% of the ventricle (Poss et al., 2002; Raya et al., 2003) (Figure 1). The ease of regeneration after resection prompted researchers to ask if other types of injury, such as ischemic cell death, would elicit the same regenerative response in this animal model. To answer this question, two additional methods to provoke heart injury in the zebrafish have recently emerged: genetic ablation of CMs (Wang et al., 2011) and cardiac cryoinjury (Chablais et al., 2011; Gonzalez-Rosa et al., 2011; Schnabel et al., 2011). Genetic ablation is based on inducible expression of the cytotoxic diphteria toxin A chain (Wang et al., 2011). This system allows ablation up to 60% of CMs, without affecting the viability of epicardium and endocardium. In cardiac cryoinjury, a nitrogen cooled probe is applied to the heart, inducing injury in 20-30% of the ventricle, damaging endocardium, myocardium and epicardium. All methods accomplish cardiac regeneration, but differ in the time needed for complete restoration of the myocardium. Complete regeneration upon ventricular resection is accomplished within 60 days, but 130 days are needed after cryoinjury (Poss et al., 2002; Gonzalez-Rosa et al., 2011). Interestingly, genetic ablation experiments lead to the fastest regeneration, 30 days being enough to recover from loss of 60% of the original CM (Wang et al., 2011). Another important difference is the amount of fibrotic tissue deposited in the injury response. While genetically induced CM death does not trigger deposition of collagen matrix and only a fibrin clot forms after resection, regeneration after cryoinjury is preceded by massive fibrosis, forming a scar-like ring around the injured area. Surprisingly, this scar does not inhibit regeneration. Indeed, a transient scar might even be necessary for heart regeneration, since prevention of scar formation by inhibiting the tgf-beta/Activin singalling pathway after cryoinjury blocks myocardial regeneration (Chablais and Jazwinska, 2012). The presence of a scar, which is eventually eliminated, suggests that the zebrafish activates mechanisms to degrade the fibrotic scar that do not operate in the hearts of adult mammals.
As a first event following injury, cardiac cells reactivate genes expressed during embryonic heart development (Kikuchi and Poss, 2012) (Figure 2). The endocardium and epicardium begin to proliferate. The epicardium forms a thickened layer covering the injured heart, a process involving epithelial-to-mesenchymal transition (EMT) of epicardial cells driven by FGF and PDGF (Lepilina et al., 2006; Kim et al., 2010). Later, proliferating CMs appear, particularly at sites close to the injury. Retinoic acid, which is synthesized by epicardium and endocardium, has been suggested to play an important role in the promotion of CM proliferation (Lepilina et al., 2006; Kikuchi et al., 2011b). The epicardium has also been proposed to guide CMs during cardiac regeneration. Epicardial cells express the cytokine cxcl12a after injury (Gonzalez-Rosa et al., 2012; Itou et al., 2012b) and treatment of inhibitors of its receptor cxcr4 impede proper regeneration of the myocardium (Itou et al., 2012b). Lineage tracing has revealed that these CMs derive from preexistent CMs (Jopling et al., 2010; Kikuchi et al., 2010). Surprisingly, regenerative potential does not decrease with age (Itou et al., 2012a). The mechanisms through which CMs reenter the cell cycle and the extent to which they need to dedifferentiate before they can undergo mitosis remain important unanswered questions. Among the genes shown to positively regulate regeneration are mitotic spindle checkpoint msp1 and polo like kinase 1, while CM proliferation is negatively regulated by p38 MAPK and miR-133 (Poss et al., 2002; Jopling et al., 2010; Jopling et al., 2012b; Yin et al., 2012). Similarly, it remains to be established if there are niches within the heart harboring CM subpopulations with different regenerative potential. Recent reports suggest that subepicardial cortical CMs might be the main contributor to the regenerating heart: clonal analysis and gata4-lineage tracing studies after ventricular resection have found a high contribution of these CMs to the regenerated ventricular apex (Kikuchi et al., 2010; Gupta and Poss, 2012).
Cardiomyocyte turnover in the mammalian heart during homeostasis and in response to injury
In mammals, cardiomyocyte cell death, as occurs after a myocardial infarction, leads to the formation of an irreversible scar. The scar is formed by proliferation of myofibroblasts, which secrete large amounts of collagen and other extracellular matrix proteins (Jennings et al., 1990; Frangogiannis, 2008). Myofibroblasts can derive from intracardiac fibroblasts or the epicardium; a contribution from blood-derived pericytes and endothelial cells has also been described (Fan et al., 2012). The scar becomes gradually more rigid and acellular, and replaces the lost myocardium. While preventing cardiac wall rupture, cardiac fibrosis brings important negative consequences such as electrical uncoupling of the remaining myocardium and the generation of arrhythmias. Persistent fibrosis in the periphery also contributes to remodeling of the ventricular geometry and the generation of arrhythmias, which can ultimately lead to heart failure.
Many attempts have been made in the past decades to detect CM proliferation in mammalian animal models as well as in the human heart (Laflamme and Murry, 2011; Sedmera and Thompson, 2011). One difficulty has been to distinguish individual CMs in tissue sections, since they are in tight contact with each other and with surrounding endocardial cells and fibroblasts. In addition, the DNA content of CMs is variable, both between and within species. In the mouse, 25% of CMs in the adult heart are mononuclear and the rest binuclear. In humans, most CMs are mononucleated (75%) but polyploid (4n), and polyploidy can increase with age up to 8n. Therefore, it is important not only to be able to tag individual CMs for a correct study of proliferation, but also to know the exact DNA content to avoid confusion between DNA duplication and real cytokinesis. Some reagents for measuring proliferation, such as labeled ribonucleotide analogs, can additionally lead to confusion of DNA repair with proliferation, labeling as “proliferative” cells that are in fact damaged and have activated their repair machinery.
Given these considerations, it is unsurprising that reports on CM proliferation in the adult human heart show wide discrepancy in the numbers obtained (for a review see (Laflamme and Murry, 2011). For example, in the report by Dr. Frisen and collaborators, 14C dating was used to estimate the age of CMs at the time of death (Bergmann et al., 2009). Their results suggest a turnover close to 50%, meaning that about half the CMs present at the time of death were present from birth, but that the rest had been added postnatally. Other reports suggest an even higher CM turnover in humans, with complete CM replenishment several times in the course of a lifetime (Kajstura et al., 2010a; Kajstura et al., 2010b). Recent progress toward a precise determination of the rate of CM turnover indicates that proliferation in the healthy human heart is mostly restricted to the first two decades of life, with approximately 0.01% of CMs proliferating in 0-to-1 year olds, dropping to fewer than 0.005% in people aged between 10 and 20 years (Mollova et al., 2013). While current estimates of the contribution of proliferation by pre-existent CMs to cardiac growth in the adult human vary, there is agreement that a certain degree of CM turnover does occur. The postnatally generated CMs have been proposed to derive from cardiac stem cells (CSC) (Kajstura et al., 2010a). However, more recent reports present evidence for CM division in the human heart, suggesting that pre-existent CMs might be the source of newly formed CMs (Mollova et al., 2013).
In the mouse, recent studies suggest that pre-existent CMs, not cardiac stem cells, are the source of newly generated CMs, during both normal homeostasis and repair (Senyo et al., 2013). Lee and collaborators combined genetic lineage tracing of pre-existent CMs with multi-isotope imaging mass spectrometry (MIMS), which allows high resolution monitoring of DNA synthesis using 15N isotope incorporation. Use of this technique to quantify CM turnover detected division of CMs during normal aging and in the response to injury, and these divisions accounted for the small increase in total CMs seen in both events. During aging in the healthy mouse, a constant birthrate of 0.76% of CMs per year was found. Division rates increased significantly 8 weeks after MI, reaching 3.2% close to the injured region.
While CM proliferation in the adult mouse heart is low, and does not lead to regeneration, during early postnatal life the hearts of rodents can regenerate after injury (Robledo, 1956; Porrello et al., 2011) (Figure 3). This regenerative capacity is rapidly lost, and if resection is performed on or after postnatal day 7, an irreversible scar is formed, as occurs after MI in the adult. Lineage tracing analysis strongly suggests that, similar to reports in zebrafish, regeneration occurs through proliferation of preexisting CMs. However, even though most CMs were labeled, at 21 days after resection 30% of the CMs present in the regenerated heart could not be traced as CM derived, leaving open the possibility that an alternative cell progenitor source is active. Indeed, a more recent report suggests that c-kit-positive cardiac progenitor cells contribute to CM regeneration after heart resection in neonates (Jesty et al., 2012). However, adult cardiac c-kit-positive cells are unable to regenerate CMs, suggesting that the differentiation potential of the c-kit cell pool is lost during aging. Supporting this conclusion, c-kit positive cells have been found to have an important paracrine role, promoting de novo formation of CMs in response to injury but not representing a stem-cell source for CMs (Loffredo et al., 2011).
Comparative analyses of the role of epicardium during cardiac regeneration
The epicardium is one of the first layers to respond to cardiac injury. During development, the epicardium contributes cells that form the coronary vasculature (endothelial and smooth muscle cells), as well as interstitial fibroblasts (Perez-Pomares and de la Pompa, 2011; Ruiz-Villalba and Perez-Pomares, 2012; Schlueter and Brand, 2012). Significantly for cardiac regeneration, EPDCs have also been reported to contribute to a cardiac mesenchymal stem-cell pool and to a small population of cardiomyocytes (Cai et al., 2008; Zhou et al., 2008; Chong et al., 2011). In the mouse, several Cre-lines have been used to study the fate of epicardium-derived progenitor cells (EPDCs) during cardiogenesis, including Wilms tumour 1- (Wt1), Gata5-, T-box transcription factor gene Tbx18-, Semaphorin 3D-, Scleraxis- , Tcf21- and PDGFR-alpha- Cre-lines (Merki et al., 2005; Cai et al., 2008; Zhou et al., 2008; Acharya et al., 2011; Chong et al., 2011; Katz et al., 2012; Rudat and Kispert, 2012; Zhou and Pu, 2012). The results of these studies present a somewhat inconsistent picture, differing in the percentage contributions to the various derivatives. These disparities can be partially explained by differences in Cre recombinase activity between the models (Rudat and Kispert, 2012). Moreover, the promoters used are not exclusive to the epicardium and thus might report transient activity in other cardiac tissues, making it difficult to accurately fate map of the epicardium based on expression of a particular gene (Christoffels et al., 2009). It is also important to remember that the epicardium is a heterogeneous cell population (Ruiz-Villalba et al., 2013), making it difficult to trace all EPDCs with a Cre-lox system based on a specific promoter. While cleary an important source of progenitor cells for the coronary vasculature and intracardiac fibroblasts, the epicardium is also important for the proliferation, maturation and compaction of the underlying myocardium, processes that are altered in mutants that affect the formation of the epicardial layer, such as Wt1-null mice (Kreidberg et al., 1993). In the healthy heart, the epicardium is a unicellular mesothelial layer covering a small underlying subepicardial space that accumulates extracellular matrix, mesenchymal cells and coronary vessels. Upon injury, epicardial cells undergo EMT, producing a thickened layer covering the injured area. As in the zebrafish, heart injury in the mouse induces the reactivation of developmental active genes (Limana et al., 2009). While it is unclear what triggers this response, reactive oxygen species released during ischemia might be involved, since hydrogen peroxide treatment can induce EMT of epicardial cells in vitro (Duan et al., 2012). The presence of a thickened epicardium has been reported in the diseased heart, in models of myocardial infarction and of pressure overload (Russell et al., 2011), suggesting a pivotal role of EPDCs in the cardiac injury response. Several lines of evidence have confirmed this hypothesis. Fate mapping of EPDCs using genetic lineage tracing of cells expressing the Wilms tumour 1 gene or Wnt 1 in mouse MI models revealed a contribution to myofibroblasts (Zhou et al., 2011a; Duan et al., 2012). However, while genetic fate mapping suggests a contribution of EPDCs to the myocardium during development, EPDCs do not significantly differentiate to CMs, after cardiac injury in the adult in any species analyzed to date (Chong et al., 2011; Kikuchi et al., 2011a; Zhou et al., 2011a; Gonzalez-Rosa et al., 2012). A role for epicardium as a source of paracrine factors is supported by experiments showing that implantation of EPDCs or the application of EPDC supernatant to the pericardial cavity of the infarcted heart improve recovery after MI (less scarring and increased ejection fraction volume) (Winter et al., 2007; Zhou et al., 2011a). Among the factors secreted by the epicardium are VEGF-A, CXCL12 and several FGFs (Zhou et al., 2011a). Interestingly, regeneration of the injured heart in the neonate is preceded by a stronger activation of Wt1 and Tbx18 expression than observed in the adult, suggesting that the increased epicardial response might be a key event allowing regeneration (Smart et al., 2011).
It is also becoming clear that the intensity and timespan of epicardium activation need to be correctly balanced for it to have a reparative effect. Consistent with this idea, administration of thymosin beta 4 rapidly activates the epicardial genes Wt1 and Tbx18, but expression drops sharply from days 4 to 7, stages at which gene expression levels remain high after severe MI (Smart et al., 2011). In accordance with this correlation, Olson and colleagues recently showed that the epicardium is a niche for neutrophils attracted by elevated RA levels after MI, and that the resulting increased inflammatory response impairs recovery upon MI (Huang et al., 2012).
In humans, published evidence suggests that EPDCs might also have an important role in cardiac repair, being a possible source of cardiac stem cells (Limana et al., 2007; van Tuyn et al., 2007; Castaldo et al., 2008)
From cardiac repair to regeneration in mammals
Why can a neonatal mammalian heart regenerate while an adult heart cannot? The reason may simply be that in the neonate the developmental program is incomplete. However, this does not fit with the massive increase in proliferation observed as a response to injury. Many more possible explanations including epigenetic changes, changes in cardiomyocyte complexity, senescence, metabolism need to be investigated in order to understand the rapid regenerative loss upon birth. Teleost fish retain capacity for growth throughout life, and a change in the density at which fish are housed can induce a rapid growth phase in which CMs are highly proliferative (Wills et al., 2008). It is thus plausible that the high regenerative capacity of the adult teleost heart arises from the fact that cells actively proliferate in these animals throughout life. Supporting this, mice overexpressing the cell-cycle regulator cyclin D2 in CMs recover better after MI than wildtype littermates, suggesting that mammalian CMs can be forced to reenter the cell cycle and divide (Hassink et al., 2008). Another consideration is the low pressure a zebrafish heart has to deal with. An adult ventricle at systole exerts a ventricular pressure of 2.5 mmHg, around 50 times less that the left ventricular pressure of a human heart (Hu et al., 2001). The high workload of the mammalian heart may make a response to injury beyond rapid tamponade with a resistant scar impractical. Zebrafish hearts are also highly trabeculated and have a very thin compact layer compared with mammals. The number of intracardiac fibroblasts is presumably very low (Ausoni and Sartore, 2009), and this might correlate with less rigid matrix, allowing easier remodeling after injury. Zebrafish are moreover very resistant to hypoxia, and activate mechanisms to adapt cardiac function to chronic hypoxia (Marques et al., 2008). While rapid re-oxygenation can prevent CM loss after an ischemic episode, analysis of the role of hypoxia in zebrafish cardiac regeneration suggests that low oxygen levels are required for CM proliferation, an observation that might have implications for clinical post-MI treatments (Jopling et al., 2012a).
Several strategies can be envisaged for the induction of cardiac regeneration in mammals, including stimulation of endogenous CM proliferation, administration of progenitor cells or differentiated CMs and their coupling to the injured heart, or differentiation of resident progenitor cells or transdifferentiation of other cardiac cell types to CMs.
Stimulation of CM proliferation
A miRNA screen has identified a subset of miRNAs that induce proliferation of neonate and adult CM both in vitro and in vivo (Eulalio et al., 2012). In addition, miR-15 has been shown to be upregulated after the first postnatal week in mice, correlating with the loss of regenerative capacity. Administration of locked nucleic acid (LNA)-modified anti-miR15 in the young and adult mouse leads to increased recovery after MI and an increase in CM proliferation (Porrello et al., 2012). MicroRNA and anti-miRNA treatments thus represent a possible strategy to promote cardiac regeneration from preexistent CMs in vivo (van Rooij and Olson, 2012). The zebrafish offers the possibility of further high-throughput screens to identify compounds that induce CM proliferation. A recently established transgenic line allows the CM cell cycle to be monitored in vivo, and screens with this line have identified a pro-regenerative role of Sonic Hedgehog (Choi et al., 2013).
Stem cells and tissue engineering strategies
In addition to strategies to activate proliferation of preexistent CMs, several progenitor cell sources for CM replenishment in the mammalian adult heart have been investigated, including bone-marrow-derived cells and resident cardiac stem cells. Cardiac stem cells can be divided into several cell types such as Isl1-positive cells, c-kit-positive cells, Sca-1-positive cardiosphere-derived cells and the Side population (Hou et al., 2012). Intensive research is being undertaken to define the contribution of these cells types to cardiac regeneration, and results to date indicate that their contribution is a combination of paracrine effects, cell fusion and transdifferentiation into CMs. Bone-marrow-derived cells seem to be involved primarily in paracrine stimulation of cardiac regeneration (e.g. (Loffredo et al., 2011). Moreover, recent reports seem to exclude a central role of Isl1 progenitor cells in CM replenishment after injury (Weinberger et al., 2012).
Clinical studies in humans have mostly focused on stem cell therapy. Some trials have yielded promising results, such as the CADUCEUS and the SCIPIO trials, using cardiosphere-derived autologous stem cells and c-kit+ cardiac stem cells, respectively (Chugh et al., 2012; Makkar et al., 2012). Nonetheless, larger cohorts of patients need to be analyzed and the long-term effects of cell therapy still need to be evaluated. Advances in tissue engineering are improving methods to deliver not only stem cells but whole tissue patches, mixed with growth factors to improve endogenous regeneration (Fleischer and Dvir, 2012). One caveat in cell therapy and tissue engineering, however, is the possible risk of arrhythmias following transplantation of ectopic cells and the lack of knowledge about how to ensure their correct electrophysiological integration. In a recent advance in this area, human embryonic-stem-cell-derived CMs (hESC-CM) grafted into a guinea pig model successfully met the physiological criteria for true mechanical and electrical coupling and functional regeneration, consolidating ESCs as one avenue towards cardiac regeneration (Shiba et al., 2012). Clinical trials using cardiac stem cells have shown clear improvements in cardiac function, but doubts remain about the contribution of delivered stem cells to myocardial regeneration.
Another cell type to be considered as an endogenous source of CM progenitors is the epicardium. Genetic fate mapping studies using Wt1 and Tbx18 Cre mouse lines revealed a small contribution to CM during development, raising the possibility that EPDCs might contribute to CMs in a similar manner in the adult. Although EPDCs usually give rise to myofibroblasts, epicardial cells can be primed to differentiate into CMs through administration of thymosin beta 4 (Smart et al., 2011). Unfortunately, this strategy worked only if thymosin beta 4 was administered before the injury, but not as a post-MI treatment, which clearly limits its clinical potential (Zhou et al., 2011b). In addition, a PDGFRalpha+ cardiac mesenchymal stem cell population derived from the proepicardium has been shown to be able to differentiate into CMs when transplanted into an infarcted mouse heart (Chong et al., 2011). This finding further supports the notion that EPDCs might be stimulated to activate a cardiogenic program.
Since the breakthrough discovery by Yamanaka enabling the transdifferentiation of somatic cells into pluripotent stem cells (Takahashi and Yamanaka, 2006), modifications to that protocol have made it possible to add a specific combination of transcription factors to promote induction of a CM phenotype (Yoshida and Yamanaka, 2011). Pioneering this field, the group of Dr. Srivastava showed that transfection of the genes encoding the transcription factors Gata4, Mef2c and Tbx5 (GMT) was sufficient to convert mouse fibroblasts into functional CMs in vitro (Ieda et al., 2010). The same team next showed that viral administration of GMT factors was able to efficiently reprogram cardiac fibroblasts in vivo (Qian et al., 2012). The efficiency of this gene therapy strategy was increased by administration of thymosin beta 4. In parallel, Dr. Olson’s laboratory obtained similar results, adding Hand2 to the GTM mix (Song et al., 2012). MicroRNA strategies represent a new path toward cardiovascular disease therapies (Quiat and Olson, 2013), and a miRNA cocktail has been identified (miRNAs 1, 133, 208, and 499) whose administration might represent a useful alternative to GMT factor gene therapy (Jayawardena et al., 2012).
Figures and Legends
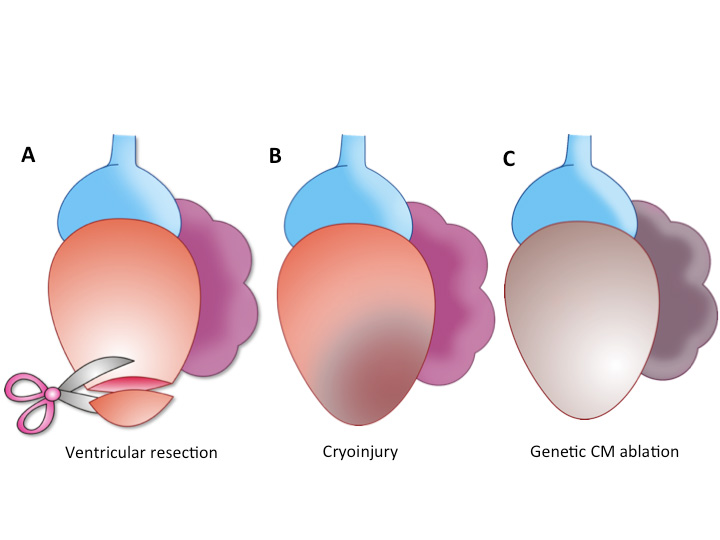
Figure 1. Models used to study heart regeneration in the zebrafish.
Current models to study mechanisms of cardiac regeneration include resection of the ventricular apex (A), ventricular cryoinjury (B) and genetic ablation of cardiomyocytes(C). As a first injury response, all of them lead to the reactivation of developmental genes and regeneration has been observed in all of them. However, the models differ in the amount of transient fibrotic tissue accumulation and in the time-span required to reach a fully regenerated heart.
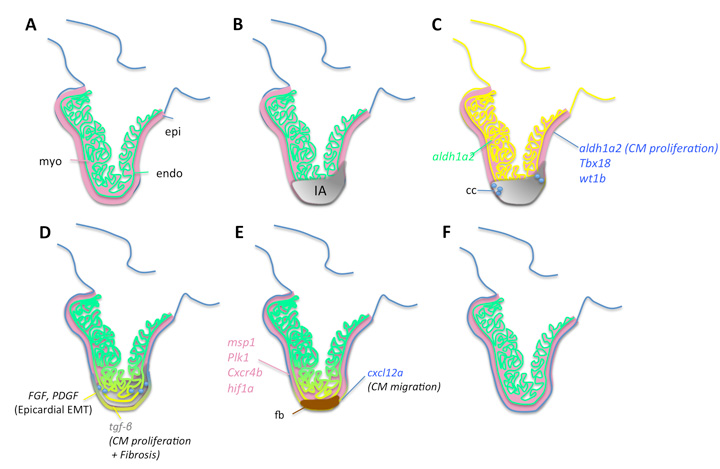
Figure 2. Events leading to zebrafish heart regeneration. Schematic illustration of the main steps leading to cardiac regeneration in the zebrafish. Some of the genes expressed during the different stages within the heart are marked as is the pathways they are controlling. A ventricle cryoinjury is represented in the model, but the scheme is aimed at summarizing all cellular and molecular mechanisms described to date independent of the injury model. (A) Sagittal section through a control heart, revealing the outer epicardial layer (blue), the inner endocardial layer (green) and the compact and trabeculated myocardium (red) of the ventricle. Injury of the ventricle; injured area (IA) is shown in grey. (B) Injury leads to expression of embryonic genes in epicardium and in the endocardium (green) as well as rapid infiltration of circulating inflammatory cells (cc) (C). (D) Epicardial cells undergo EMT and form a thickened cap covering the injured area (IA). Endocardial cells proliferate and invade the IA. (E) Fibrotic tissue deposition (fb) appears transiently (not observed upon genetic CM ablation), which becomes subsequently degraded. Finally, cardiomyocyte proliferation leads to regeneration of the missing myocardium (F).
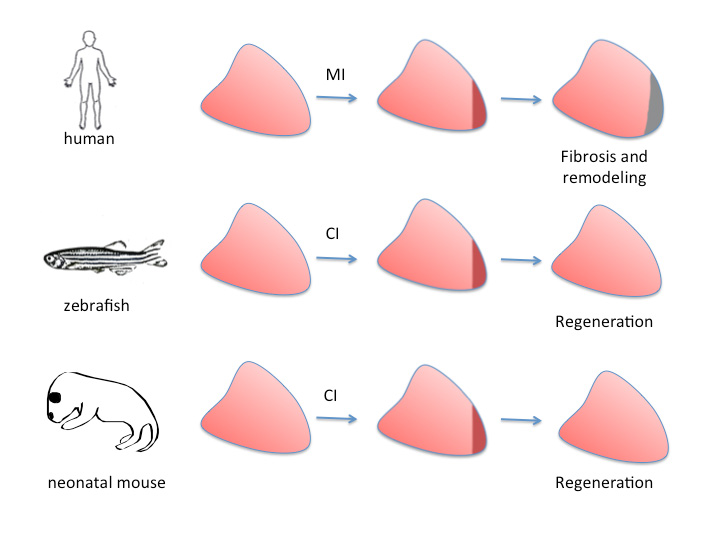
Figure 3. Different regenerative capacities of the hearts of adult humans, adult zebrafish and neonatal mice.
(A) Upon myocardial infarction (MI) in adult humans, cardiomyocytes die and are replaced by fibrotic tissue (brown) which matures into an irreversible scar (grey). In a MI model in the zebrafish (cryoinjury, CI), a transient scar is formed (brown), followed by complete regeneration. (C) Neonatal mice are also able to regenerate myocardium upon cryoinjury or ventricular resection if the injury is performed during the first week of life.
Conclusion:
Conclusion and Perspectives
Several recent discoveries—the non-postmitotic status of adult myocardium, the rapid epicardial response and the re-expression of developmental genes upon injury—point to similarities between injury response mechanisms in the hearts of lower vertebrates and mammals. There are, however, important differences. While mammalian CMs seem to react to injury by hypertrophy, zebrafish CMs undergo hyperplasia. This difference suggests halted cytokinesis in mammalian CMs, which might be possible to overcome therapeutically. Another important difference is the ability of lower vertebrate hearts to regress the fibrotic scar. This might be directly linked to the proliferative capacity of CMs, or alternatively might reflect intrinsic differences in fibroblast biology. Understanding how the hearts of lower vertebrates and neonatal mammals are able to regenerate will be invaluable for progress toward cardiac replenishment in the human adult heart. Transformation of scar tissue into myocardium, as achieved in mouse models, has major potential for therapeutic development and might be the next great challenge on the road to achieving complete cardiac regeneration in humans.