
However, the simplest hearts such as those found in tunicates (7) or Drosophila (40) resemble a pulsatile blood vessel with two layers of cells, myocardium and epicardium, and remarkably lack the endocardium. In some species, there are pulsatile structures within the lymphatic trunks that propel the lymph, named lymphatic hearts; however, they seem to be mostly related to skeletal muscle (37).
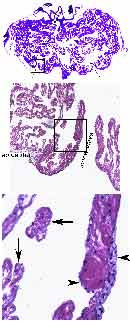
Figure 1
Development of the myocardium starts during gastrulation with the precardiac mesoderm, organized into cardiac crescent (15). Anteriorly localized cells would normally give rise to the left ventricle, while posteriorly located ones would mostly contribute to the atrial tissues; however, their fates are not set in stone and are plastic in response to local environment cues. There is also the second, by some called secondary or anterior, heart field, which would give later on rise to the right ventricle and outflow tract (see Robert Kelly’s text on outflow tract development on this site, ).
As the cells migrate towards the midline, they start to organize themselves into two tubular structures with presumptive endocardium on the inside and myocardium on the outside. In the midline, the fusion process starts first with the endocardial tubes, and later on with myocardium. If this fusion process is perturbed (mechanically, or by retinoid deficiency (16)), an embryonic lethal malformation known as cardia bifida (Figure 2) results.
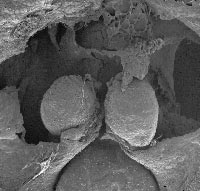
Figure 2
The primitive cardiac tube is initially straight, and is composed, from outside to inside, of myocardium, acellular cardiac jelly, and endocardium. It contains the portion that will become the left ventricle and presumptive atria are located at the inflow (posterior) end. The first contractions commence in the ventricular segment, and soon, the pacemaking area is localized at the inflow (venous) pole (9, 13) and circulation of blood starts.
The crucial process for further morphogenesis and septation of the heart is termed cardiac looping. In a strict sense, the initially straight tube is thus transformed into a cardiac loop, and at that period begins expansion of atrial and ventricular chambers, which acquire their molecular identity. The original (“primitive”, slowly conducting and contracting) myocardium flanks the atrial and ventricular segments, forming sinus venosus at the inflow (venous) end, atrioventricular canal between the atria and ventricle, and myocardial outflow tract at the outflow (arterial) end. This process is well described from morphological point of view by Manner (19), and the molecular fingerprints of chambers are continuously updated by the Moorman lab (24).
From a clinical standpoint, people are most interested in further morphogenesis of chamber myocardium, since its architecture is an important determinant of heart pumping function (27). In the atrial chambers, two compartments can be distinguished: the original embryonic atrium, which contains the pectinate muscles that vary in form and extent considerably among species (31, 33), and outline the atrial appendages or auricles. Their complex labyrinth can be a site of thrombus formation due to slowing of blood flow, and is used clinically for wedging the atrial pacing electrode (in the right atrial appendage, which is considerably larger in humans and this difference also defines the morphological laterality of the atrial chambers). The pectinate muscles appear slightly later (31, 33) than ventricular trabeculae, described in detail below.
Formation of trabeculae in the ventricular chambers is the hallmark of their differentiation and distinguishes these segments morphologically from the rest of the looped heart, where the myocardium is organized into a circular, multilayered tube, resembling tunica media of the blood vessels. Ventricular trabeculae express different patterns of genes, such as connexin40 (3, 23) that contributes to its higher conduction velocity. A molecular pathway considered responsible for trabeculation is the neuregulin pathway (8, 17, 21), but lack of trabeculation together with early (~ED10) embryolethality was reported in other null mice as well (for review, see (39)). It is believed that the primary reason for trabeculation is the necessity to provide sufficient nutrition for increased myocardial mass in the (initial) absence of coronary circulation (22). Indeed, the ventricles of some adult lower vertebrates are organized in such way (36), even though there are coronary arteries present in different parts of those hearts (32). Such trabeculated hearts work well as volume pumps, but are ill-suited for generating higher pressure necessary for sustained aerobic performance. Not surprisingly, a different morphological form of the ventricle, dependent primarily on the thick, vascularized outer compact layer, is found in large, fast-swimming fish species (1, 26).
During ontogenesis of higher vertebrates, compaction of the trabeculae coincides with the functional deployment of coronary circulation (Figures 3, 4, 5, (31)). The importance of this process is emphasized by ~ED14 lethality of mouse mutants with generalized failure of compaction (reviewed in (39)). It should be noted, however, that there are other cardiac causes of embryonic lethality, such as grossly abnormal cushion or valve development, that are only recently being revealed due to advances in non-invasive prenatal imaging (25, 28). This “ventricular non-compaction”, by some called “the puny heart syndrome” is associated with deletion of genes from different pathways, from transcription factors (WT1) through retinoid receptors (RXR) to vasculogenic molecules (VCAM). It is possible that the similarity of these phenotypes is only superficial, and detailed comparative morphological and molecular examination would reveal subtle yet potentially important differences. All these mutants are, however, significantly different from recently recognized human condition called isolated left ventricular non-compaction (6, 10, 38), or non-compaction cardiomyopathy, which is localized rather than general, and typically leads to cardiac failure during postnatal life. Details of this cardiomyopathy and its possible developmental origins are detailed below.
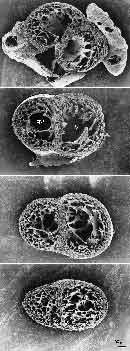
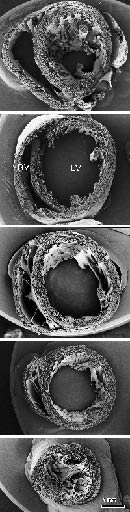
Figure 3 Figure 4
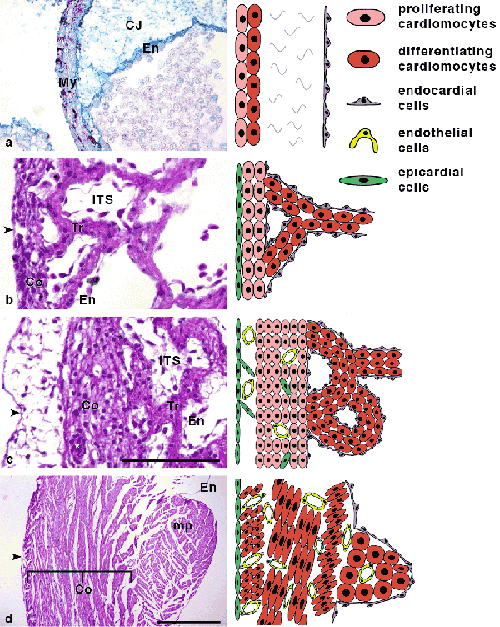
Figure 5
Development of the heart of higher vertebrates has in recent years received considerable attention, primarily in the context of pathogenesis of congenital heart defects, which are the most common congenital anomaly (~1%). Most studies are focused on cardiac (mal) septation, since this is the prevalent form of CHD. Primary non-ischemic diseases of the myocardium are rare, and apart from tumors, myocarditis or secondary metabolic disorders, are called cardiomyopathies. There are different ways to classify them, but most common division is into hypertrophic, dilated, arrhythmogenic, and non-compacted. Sometimes the boundaries are not quite distinct, and manifestations of the same gene mutations (which now seem to be the underlying cause of most cases) can change in time (6, 14). More longitudinal information from clinical studies outlining the natural history of these diseases will be necessary before we will be able to decide if these conditions are indeed congenital, and relate them in any way to described models of mouse trabecular non-compaction.
Even after compaction, the structure of the compact myocardium develops further, primarily by increasing the number of myocyte layers and the spiraling of their course (27, 29). This process is, at least in part, regulated by functional loading, as was shown by studies in chick embryos subjected to altered loading conditions (30, 35). Increased pressure loading leads not only to premature compaction and spiraling of trabeculae, but also to accelerated development of a transmural gradient in fibre inclination in the compact myocardium. Conversely, the hypoplastic left ventricle presents a more immature pattern of myofiber orientation. This process is spread over a longer time period in humans, occurring during the second trimester of gestation (11, 12). It is believed that such three dimensional architecture (2) is important for ventricular contractile function, and can be one reason for late failure of univentricular hearts with a single pumping chamber of right ventricular morphology in the setting of Fontan circulation (18, 34).
Figure legends
- Figure 1. Complex heart morphology of some marine invertebrates. The myocardial architecture of blue crab heart is highly trabeculated and inflow (top panel, arrows; higher power view shown in the middle panel) as well as the outflow (arrowhead) valves are well developed; however, there is no endocardium covering this myocardial labyrinth (arrows in high power view, bottom panel) in contrast to probably epicardially-derived covering of inflow valve leaflets (arrowheads in the bottom panel).
- Figure 2. Cardia bifida in vitamin A-deficient quail embryo (courtesy of Maia Zile) in scanning electron micrograph.
- Figure 3. Pre-compaction mouse heart (ED 13.5) in transverse sections. These SEMs were prepared in the Pexieder lab at Universite de Lausanne by the late Dr. Si Minh Pham. LV, left ventricle, RV, right ventricle.
- Figure 4. Compact myocardium of the adult mouse in transverse sections. Compare with Figure 3. Abbreviations as above.
- Figure 5. Trabecular emergence and compaction. Note the transition from nutrition by luminal diffusion to capillary bed. From (31).