Calcium (Ca2+) is a unique second messenger that translates electrical signals into mechanical activity in the cardiomyocyte. Physiological excitation-contraction coupling (ECC) is a highly coordinated process which is characterized by a well-defined, triggered Ca2+ release out of the sarcoplasmic reticulum (SR) in systole followed by a quick Ca2+ elimination out of the cytosol in order to induce relaxation of the myofilaments in diastole.
This process includes a number of transporters, ion channels and pumps which contribute to highly regulated Ca2+ fluxes on a beat-to-beat basis.
Voltage gated Na+ channels are activated upon depolarization of the cell membrane, which leads to a rapid influx of Na+ generating the fast upstroke (phase 0) of the cardiac action potential (AP). This leads to a steep increase of the membrane potential raising the probability that the voltage dependent L-type Ca2+-channels will open. As a consequence, Ca2+ passively drawn by a concentration and electrochemical gradient enters the cardiomyocyte, thereby producing the plateau phase of the AP. Ca2+-ions bind to ryanodine receptors type 2 (RyR2) which are located on the surface of the SR and trigger the release of an even bigger amount of Ca2+ out of this intracellular Ca2+ storage. This mechanism of Ca2+-induced Ca2+-release leads to a prominent increase in cytosolic Ca2+ which binds to troponin C (TnC) and thereby induces actin-myosin interaction. For cardiac relaxation, Ca2+ needs to be eliminated from the cytosol. The SR Ca2+-ATPase (SERCA) and the Na+/Ca2+-exchanger (NCX) are the main mechanisms for Ca2+ removal. Ca2+ removal is mainly achieved by an active, energy-consuming Ca2+-reuptake into the SR via SERCA2a. A smaller amount (in humans ~30%) of Ca2+ is extruded out of the cell via the NCX which is a facilitated diffusion whereby the electrochemical potential gradients of Na+ and Ca2+ are the source of energy to drive the transport. This ion exchanger produces an electrical current because 3 Na+ are exchanged for 1 Ca2+ and can thus induce arrhythmogenic triggers (transient inward current).
Since cellular Ca2+ concentrations make up the molecular trigger for cardiac contraction and relaxation, it is a matter of course that this system is elaborately regulated and highly adaptable to physical demands and hence needs several regulator mechanisms for fine tuning. SERCA2a activity and hence speed of relaxation is influenced by phospholamban (PLB) that is one of the major mediators of the cardiac contractility response upon β-adrenergic stimulation. Inhibition of SERCA2a by PLB is dependent on the phosphorylation status of PLB and is most pronounced when PLB is unphosphorylated. Intracellular protein kinases such as protein kinase A (PKA) as well as the Ca2+/calmodulin-dependent protein kinase IIδ (CaMKIIδ) can phosphorylate PLB at specific residues and thereby abandon its inhibitory effect on SERCA2a. A second subcellular microdomain is the RyR2-complex that consists of several proteins and whose Ca2+ release capacity and diastolic closure can be regulated via phosphorylation by PKA and CaMKII.
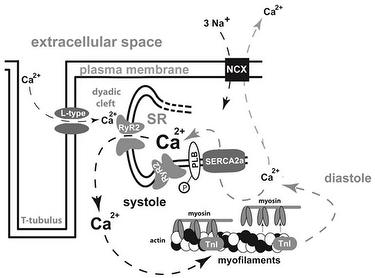
Figure legend: Mechanisms of excitation–contraction coupling in a cardiomyocytes; arrows indicate Ca2+ shifts in systole (left) and diastole (right); L-type L-type Ca2+ channel; RyR2 ryanodine receptor type 2; SR sarcoplasmic reticulum; PLB phospholamban; TnI troponin I; NCX Na+–Ca2+-exchanger; SERCA2a sarcoplasmic endoplasmic reticulum Ca2+- ATPase 2a; P phosphate.
Modified from Fischer et al., Heart Fail Rev 2013.