
Background
Inherited arrhythmogenic diseases constitute an important cause of sudden death (SCD). They affect mostly young and otherwise healthy people and have been considered one of the most common causes of SCD in young athletes (1). These conditions include long QT syndrome (LQTS), Brugada syndrome, catecholaminergic polymorphic ventricular tachycardia and, more recently, short QT syndrome (SQTS) (2).
SQTS is a cardiac channelopathy characterized by an abnormally short QT interval and an increased risk of atrial and ventricular fibrillation. Although the link between a short QT interval and sudden cardiac death had been previously suspected (3), the first clinical cases were reported during the last decade (4,5) and the knowledge about this syndrome has significantly increased since then.
Mechanism
As in other inherited arrhythmogenic diseases, SQTS has been related to several mutations affecting the function of ion channels responsible for the currents that generate the cardiac action potential. Several mutations may cause either hyperfunction of the delayed rectifier potassium current or hypofunction of the calcium current (figure 1). These result in a shortening of the repolarization period and an increase in transmural dispersion of repolarization which explain the main features of this syndrome: short QT interval, short atrial and ventricular effective refractory periods and, as a result of them, susceptibility to atrial and ventricular fibrillation.
Figure 1. Main dysfunction of cardiac ion channels in SQTS.
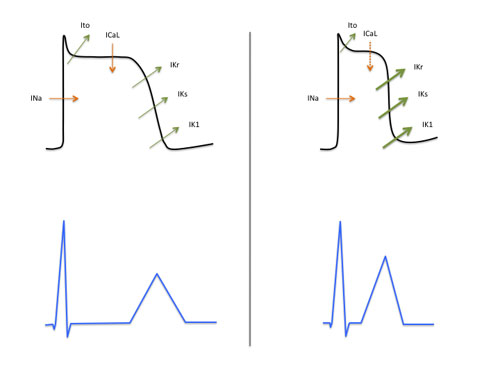
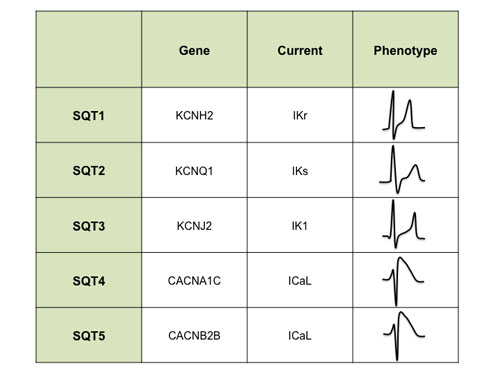
SQTS is a heterogeneous disease both from genotypic and phenotypic point of view. Five subtypes of SQTS have been described so far, which correspond to seven mutations in five different genes encoding different cardiac ion channels (table 1).
Table 1. Classification of SQTS according to genotype.
Most of them are familial cases and the pattern of inheritance seems to be autosomal dominant. Interestingly, four of these five genes have been implicated in the etiology of long QT syndrome but with mutations in the opposite sense, that’s to say, loss of function mutations instead of gain of function mutations of potassium channels.
SQTS 1 was first described in 2004 by Brugada et al (6) and is the variant present in the majority of patients. They reported gain of function mutations in KCNH2 that increased IKr current and led to marked shortening of action potential in two families with short QT interval and high incidence of sudden cardiac death.
SQTS 2 was reported in 2004 by Bellocq et al (7). They presented an alternative molecular mechanism for a patient with short QT and ventricular fibrillation: a gain of function mutation in KCNQ1 that enhanced IKs current. However, there are few and sporadic cases of this variant documented so far.
One year later, Priori et al (8) identified the third variant of this syndrome (SQT3) in two patients. A genetic defect in the KCNJ2 gene caused a significant increase in the outward Ik1 current leading to an acceleration of the final phase of the repolarization.
Finally, in 2007, Antzelevitch et al (9) described two new variants with similar channel dysfunction: loss-of-function mutations in genes encoding the α1- and β2b-subunits of the L-type calcium channel associated with a familial sudden cardiac death syndrome in which a Brugada syndrome phenotype is combined with QT intervals shorter than normal. Both mutations have been termed, respectively, SQTS 4 (2 patients) and SQTS 5 (7 patients).
Nevertheless, gene mutations have not been found in all patients with short QT syndrome and the factors responsible of the malignancy and expression of the known mutations have not been identified. This heterogeneity, together with the paucity of cases, makes this a challenging field of research.
Clinical presentation
The clinical presentation of SQTS is also diverse with a high penetrance but a great variability of expression between different families and even among members of the same family.
SQTS is characterized by a high lethality. In the largest available case series of SQTS (10), cardiac arrest was the most frequent symptom (34%) and the most frequent first clinical presentation (28%). Although it usually occurs in adults (median age 30 years), the age of presentation ranges from a few months to the sixth decade of life. Unlike LQTS, there were no specific triggers for episodes that took place at rest, during exercise, or after loud noises. Viskin et al (11) have also reported that men with idiopathic ventricular fibrillation show a QT shorter and a higher prevalence of short QT interval than healthy males.
Consequently, this syndrome has to be excluded in patients without structural heart disease presenting with sudden cardiac death.
Other symptoms often documented are syncope and palpitations. In the series of Giustetto (10), syncope was the first presenting symptom in 24% of cases and self-terminating ventricular fibrillation (VF) episodes were considered the most likely mechanism. Up to 31% of patients referred palpitations and atrial fibrillation was present in more than 80% of them despite his young age (even in adolescents and children). Atrial fibrillation constitutes one of the main findings of SQTS and therefore it is recommended to take it into account in the management of young patients with lone atrial fibrillation.
Diagnosis
SQTS diagnosis is based on the evaluation of symptoms, patient's family history and 12-lead ECG. It is essential to question the patient about the presence of key symptoms (syncope and palpitations) and family history of syncope, sudden death or atrial fibrillation at a young age. Secondary causes of short QT must also be excluded and include hyperthermia, hyperkalemia, hypercalcemia, acidosis and alterations of the autonomic tone.
When evaluating the ECG (figure 2) in these patients, three main aspects need to be considered: the duration of the QT interval, the morphology of T wave and the behaviour of both of them with the heart rate.
Figure 2. ECG taken from a patient diagnosed with SQTS: QTc 320 ms
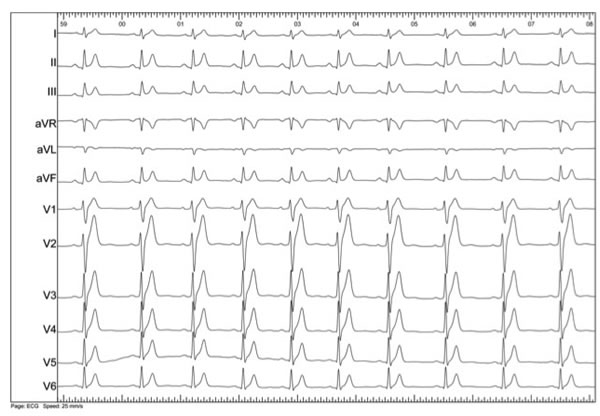
As in LQTS, there is not a single QTc value to differentiate most cases of SQTS from healthy individuals. In the first published series (6-8), patients presented QTc with values shorter than 300-320 ms, while in genotypes more recently described9 (SQTS 4 and 5), they were just shorter than 360 ms. As recently reviewed by Viskin (12), males with QTc <330 ms and females with QTc <340 ms should be diagnosed with SQTS even if they are asymptomatic since this values are very rare in healthy population. In addition, QTc intervals shorter than 360 and 370 ms (males and females respectively) should only be considered diagnostic of SQTS when supported by symptoms or family history because they overlap with healthy population.
It is not only important to assess the value of QT but also its accommodation to heart rate. Patients with SQTS show constant QT values and a lack of adaptation to heart rate with failure to prolong adequately at slower heart rates and abnormal shortening during acceleration (pseudonormalization of the QT interval at rapid rates). Serial ECGs, Holter monitoring and treadmill testing may be useful for a correct diagnosis and prevents unrecognition of patients with an elevated heart rate at baseline. In addition, they can reduce wrong diagnosis in SQT patients with sinus bradycardia since it is known that Bazett formula overcorrects the QT interval at slow heart rates.
As for the morphology of ST segment, SQTS patients share a short or even absent ST segment, with the T wave initiating immediately after the S wave. T wave is usually taller and narrower than in normal subjects. Recently, some features of T wave have been published to distinguish SQTS from healthy subjects with short QT interval. Anttonen et al (13) reported that patients with symptomatic SQTS patients have significantly shorter Jpoint-Tpeak interval and frequently shorter Tpeak-Tend intervals. Watanabe et al (14)observed that early repolarization was more common in SQTS patients (65%) and, moreover, that it was associated with the appearance of arrhythmic events. They also corroborate that duration from T-wave peak to T-wave end was longer in these patients. T wave features can also guide about the patient’s genotype. SQTS 16 often have tall, peaked and symmetrical T waves, SQTS 38 show asymmetrical peaked T wave due to the acceleration of the final phase of the action potential repolarization, and SQTS 4 and 59 coexist with Brugada type ST elevation in precordial leads V1 and V2 at baseline or after administration of ajmaline. Nevertheless, these data should be interpreted carefully given the small number of patients reported so far, especially in some genotypes.
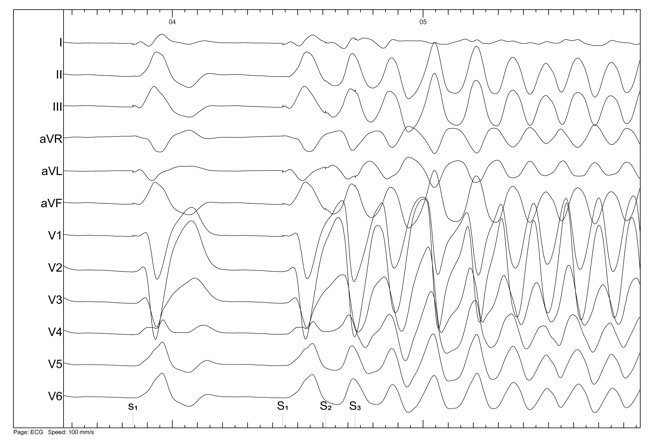
The role of electrophysiological testing in patients with SQTS remains controversial. Some studies showed very short atrial and ventricular effective refractory periods, high rate of inducible atrial and ventricular fibrillation, and a marked vulnerability to mechanical induction of ventricular fibrillation (figure 3 to 5). However, in the study of Giustetto et al10, the sensitivity of electrophysiological study for detection of vulnerability to ventricular fibrillation was 50% (three patients out of six with documented ventricular fibrillation), some case reports showed that noninducibility does not exclude future ventricular fibrillation episodes (15) and the clinical significance of inducible VF in asymptomatic patients is unknown. Therefore, it is uncertain how useful this test could be in the diagnosis and stratification of these patients.
Figure 3. Ventricular effective refractory period shorter than 400/140 ms.
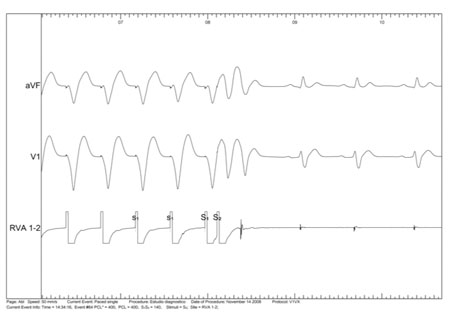
Figure 4. Atrial effective refractory period shorter than 400/200 ms.
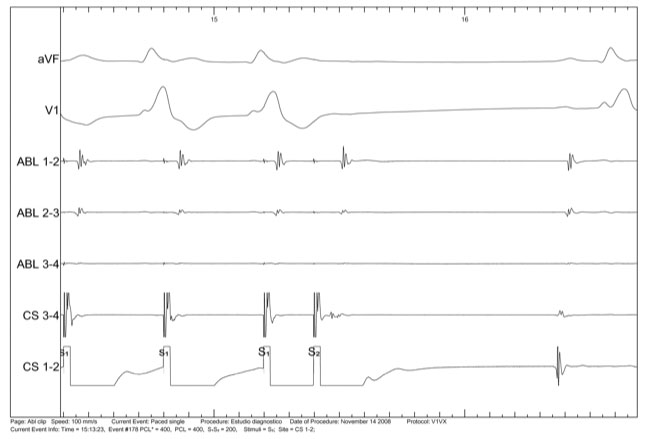
Figure 5. Induction of ventricular fibrillation during ventricular programmed stimulation in a patient with SQTS
The contribution of genetic testing is not clearly defined either. To date, seven genetic mutations have been identified but a correlation between genotype and phenotype does not yet exist due to the relatively few cases described and genetically confirmed. Furthermore, not all mutations are known. Isolated reported cases (16) suggest that it may facilitate appropriate decision-making in symptomatic patients with an abnormal but nondiagnostic ECG given the prognostic and therapeutic implications. However, a negative test does not rule out the syndrome, as there are mutations unidentified. It may also prove useful in family screening not only to diagnose asymptomatic carriers at an early stage but also to definitively identify non-affected members of a certain mutation.
Management
SQTS entails a high risk of sudden cardiac death from fatal arrhythmic events and a high penetrance in affected families. Therefore, ICD is the mainstay therapy for these patients. While this is obvious in symptomatic patients, doubts may arise when dealing with asymptomatic patients, especially if they have no family history. So far, there is not enough evidence for clear risk stratification given the low number of documented cases and its relatively recent diagnosis. It is generally accepted that clinical manifestations, family history and a positive electrophysiological study or genetic test may support the implantation of an ICD. However, a negative result does not exclude the diagnosis or the possibility of future arrhythmic events.
Even though ICD is the only effective treatment, it also has specific problems in these patients. On the one hand, some reports showed an increased risk of inappropriate therapy due to sinus tachycardia, atrial fibrillation and, above all, oversensing of T wave which are often tall and narrow. In the study published by Schimpf et al (17), 3 of 5 patients received inappropriate shocks due to T wave oversensing shortly after implantation and despite no evidence of abnormalities at prehospital discharge testing. The reason was a postoperative reduction of the R wave amplitude and increase of the T wave signal. Therefore adaptation of standard programming to prevent T wave oversensing must be considered after implantation and during follow up though always ensuring an adequate sensing of ventricular arrhythmias. On the other hand, ICD implantation in children increases technical difficulties and complications and is not feasible in the youngest.
Although ICD therapy is the fist line therapy, pharmacological therapy can be indicated in some cases:
- As an alternative to ICD implantation in young children
- Patients with contraindications or declining ICD implantation
- As an adjunctive therapy to prevent appropriate discharges
- Prevention of symptomatic episodes of atrial fibrillation
However, drugs must be used with caution since the long-term efficacy of drug therapy in preventing serious arrhythmic events has been studied only in SQTS 1 patients and is not well established.
Quinidine is considered the most effective pharmacological therapy in these patients. It blocks several potassium channels (IKr, IKs, Ito, IKATP and IK1) and the inward sodium and calcium currents. In SQTS 1 quinidine has shown to produce a marked prolongation of the QT interval and ventricular effective refractory periods, prolongation of the ST segment and T wave duration, restoration of heart rate dependence of the QT interval, repolarization dispersion decrease and prevention of ventricular fibrillation induction (18-20). However, the clinical consequences of these electrophysiological effects are unknown.
Subsequent studies (21) have revealed that oral disopyramide prolongs QT interval and ventricular effective refractory periods in patients with SQTS 1 and proposed it as an alternative to quinidine.
Studies with other antiarrhythmics drugs have failed to show any beneficial effect. Initially, flecainide seemed to prolong slightly QTc interval and reduce the inducibility of ventricular fibrillation during electrophysiological study (5) but later studies have not confirmed this effect (18). Propafenone has shown to be effective in preventing frequent paroxysms of AF with no recurrence of arrhythmia for more than two years, and without any effect on QT interval (22). Ibutilide and sotalol, IKr blockers, have been demonstrated to be ineffective to prolong the QT interval (18). Wolpert et al (19) proved that this resulted from the mutation in KCNH2 reducing the affinity reducing the affinity of sotalol for the IKr channel unlike quinidine. It is possible that the advantage of quinidine over sotalol is also related to its multichannel effect.