I - Background
The Anderson - Fabry disease is a rare X-linked lysosomal storage disorder caused by a-galactosidase A deficiency. The disease leads to a progressive accumulation of globotriaosylceramide in various organ systems [1] and a wide variety of signs and symptoms including acroparaesthesiae, pain crisis, heat intolerance, hypohydrosis, gastrointestinal symptoms, angiokeratomas, corneal dystrophy, renal impairment, cerebrovascular disease and cardiomyopathy.
Heterozygous female patients are not only carriers of the disease they may also show a wide range of disease severity, ranging from a relatively benign course to manifestations comparable with those of hemizygous men [2].
Life expectancy in men is reported to be reduced by about 20 years from that of the general population and the most frequent cause of death in males is end-stage renal failure followed by cardiac involvement [3] which is characterised by a progressive left ventricular hypertrophy (LVH) mimicking the clinical phenotype of hypertrophic cardiomyopathy [4].
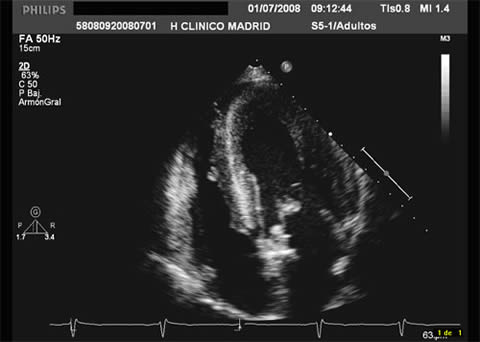
Interestingly, Fabry disease has been reported as the cause of left ventricular hypertrophy in 6% of men with a late-onset of hypertrophic cardiomyopathy. Concentric hypertrophy patterns are most frequent but asymmetric septal hypertrophy occurs in 5% of cases and dynamic left ventricular outflow obstructions are rare. As the disease progresses, interstitial abnormalities and replacement myocardial fibrosis become important. Other cardiac manifestations are valve infiltration with regurgitation and a wide variety of rhythm abnormalities.
II - Diagnosis
Diagnosis is confirmed by biochemical demonstration of a reduction in the activity of α- Galatosidase A in peripheral blood cells and the detection of casual mutation. The standard non-invasive tools for identifying patients with Fabry and heart involvement clearly are ECG, conventional 2D-echocardiography and cardiovascular magnetic resonance. ECG provides information about electrical alterations or the presence of LVH. Echo offers an estimate LV mass and assessment of LV function and possible Valvular alterations MRI can detect the preence of late enhancement with gadolinium that can be a sign of fibrosis in patients with cardiac FAbry manifestations.
This image diagnostic approach is useful in patients with established cardiomyopathy but it is not suitable to detect impaired myocardial function early in the course of the disease or sensitive enough for the screening of Fabry relatives. For early detection, tissue Doppler imaging (TDI) has emerged as a sensitive and non-invasive tool for detecting impaired contraction and relaxation myocardial function in several forms of inherited cardiomyopathies [5-8].
III - Treatment
The advent of enzyme replacement treatment (ERT) by recombinant α-galactosiadase A has increased the need for early recognition to help treatment and delay or prevent complications. Most published studies have focused on Fabry patients with advanced organ involvement which probably explains the variation in response to ERT seen in a number of them [9-10]. Current guidelines indicate that all males over the age of 16 years should be offered ERT as a strategy that would ensure the initiation of ERT before major organ damage has progressed to irreversible stages, however the recommendations for women are less clear [11]. Further studies are needed to assess the prognostic implications of ERT in patients without established cardiomyopathy and usefulness of TDI and TDI-derived techniques in clinical decision making.
Figure 1 shows slightly reduced systolic TDI velocities (7 cm/sec) in a Fabry male patient with normal parietal thickness.
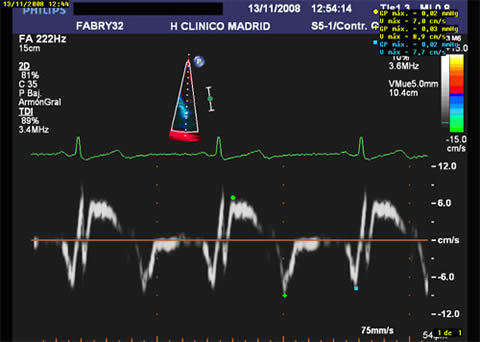
Figure 2 represents the typical phenotype of concentric LVH in a Fabry male patient.