
The risk of heart failure in diabetes
The association of diabetes with hypertension, obesity and coronary heart disease, is well established and predisposes diabetic subjects to heart failure (HF) [1]. This has been linked to duration of diabetes and the extent of glycemic control [2]. Cardiovascular morbidity and mortality in diabetes are strikingly high with a reduction in life expectancy. A recent Cambridge study has shown that individuals in their sixties who have a combination of diabetes and heart disease have an average reduction in life expectancy of about 15 years [3]. Four decades ago the Framingham study firmly established the epidemiologic link between diabetes and HF [4], showing that diabetes predicted heart failure independent of hypertension, age, obesity, dyslipidemia and coronary disease. When CHD and valve disease were excluded, the relative risk of HF remained elevated at 3.8 fold in diabetic men and 5.5 fold in diabetic women. It is now recognized that in addition to accelerating and worsening the consequences of coronary artery disease and hypertension, diabetes also has a direct effect on the myocardium, placing the diabetic subject at an increased risk of developing heart failure.
Diabetes increases the odds of a non-ischemic dilated cardiomyopathy (odds ratio: 1.75; 95% CI: 1.71-1.79), which has been associated with increased myocardial stiffness [5]. This entity of a unique diabetic cardiomyopathy (DMCMO) was originally proposed by Lundbeek [6], and subsequently confirmed at autopsy by Rubler et al in 1972 [7] in four diabetic patients who presented with heart failure (HF) and showed no evidence of hypertension (HT), CAD, or valvular disease. Rubler found evidence of myocardial hypertrophy, fibrosis, and microvascular changes in keeping with dilated cardiomyopathy. The development of myocardial dysfunction in diabetes has been attributed to the effects of lipotoxicity, microvascular AGEs deposition, microvascular rarefaction, and autoimmunity, all of which contribute to produce myocardial fibrosis and/or myocardial hypertrophy, the hallmarks of DMCMO [8] that are described below (Table 1).
Table 1. Contribution of pathophysiological mechanisms to the phenotypes of diabetic cardiomyopathy
|
Metabolic overload effects |
HFPEF |
HFREF |
|---|---|---|
|
Insulin resistance/hyperinsulinemia |
+++ |
+ |
|
Hyperglycemia |
+++ |
+ |
|
Lipotoxicity & oxidative stress |
+++ |
+ |
|
AGEs deposition |
+++ |
+++ |
|
Microvascular rarefaction |
+++ |
+++ |
|
Autoimmunity |
- |
+++ |
|
Structural changes |
Functional effects |
|
|
Stimulus to ventricular remodeling |
Endothelial dysfunction |
Autoimmune cell death |
|
Effects of microvascular dysfunction |
Low NO availability |
Hypoxic damage |
|
Cardiomyocyte changes |
Hypertrophy & stiffening |
Stiffening & apoptosis |
|
Myocardial matrix changes |
AGEs +reactive fibrosis |
AGEs + replacement fibrosis |
|
|
Diastolic dysfunction |
Systolic dysfunction |
AGEs: advanced glycation end-products; HFPEF: heart failure with preserved ejection fraction; HFREF: heart failure with reduced ejection fraction; NO: nitric oxide
Adapted from Seferovic and Paulus [11].
Pathogenetic mechanisms of myocardial dysfunction in diabetes
Role of hyperglycemia
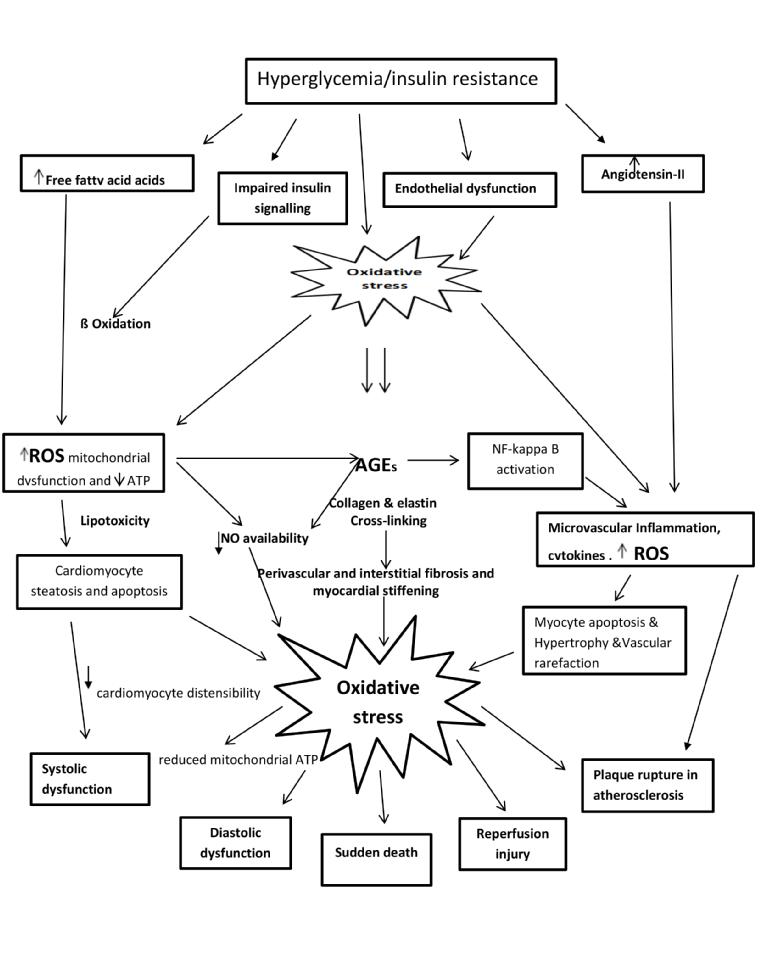
Hyperglycemia (Figure 1) increases the level of free fatty acids and growth factors in the myocardium, and causes abnormalities in substrate supply and utilization. Furthermore, it is toxic to the endothelial cell, causes mitochondrial damage [9] and induces oxidative stress and the release of superoxides, leading to abnormal gene expression, impaired production of nitric oxide (NO) and reduced distensibility of cardiomyocytes.
Hyperglycemia activates the renin-angiotensin system in myocardial cells (Figure 1) leading to cell growth and cardiac hypertrophy and these damaging effects can be further aggravated by oxidative stress. In addition, it stimulates collagen production and crosslinking as well as the production and deposition of non-enzymatic formation of advanced glycation end-products (AGEs) in the coronary microvasculature and the myocardial interstitium. AGEs trigger vascular inflammation, lower myocardial NO bioavailability and contribute to concentric LV remodeling. AGEs-induced crosslinking in collagen and elastin and result in increased myocardial stiffness and impaired cardiac relaxation in the diabetic heart that has been correlated with tissue Doppler indices of diastolic dysfunction [10].
Figure 1. Cardiometabolic mechanisms in myocyte injury.
Insulin resistance/hyperinsulinemia
Insulin resistance (IR) affects a number of signaling pathways, causing cardiomyocyte hypertrophy, reactive interstitial fibrosis and expression of myocardial titin, all of which contribute to the reduction in cardiomyocyte distensibility. Central obesity (associated with IR) and microvascular rarefaction cause the release of proinflammatory cytokines and the generation of reactive oxygen species (ROS), leading to an inflammatory state in the coronary microvasculature with a reduction in nitric oxide (NO) bioavailability, increased vessel permeability and programmed cell death (apoptosis). This coronary microvascular endothelial dysfunction is thought to drive the development of myocyte hypertrophy with concentric remodeling and myocardial stiffening resulting in LV diastolic dysfunction (Figure 2) [11].
Figure 2. Transmitral and tissue Doppler in a normal subject and in a subject with diabetic cardiomyopathy.
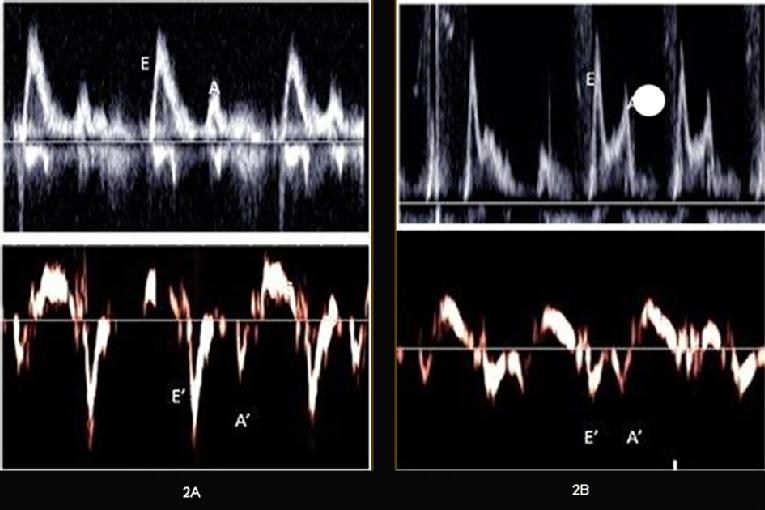
Transmitral (upper panels) and tissue Doppler (lower panels) in a normal subject (A) and a subject with diabetic cardiomyopathy (B). Notice the reduced E’ compared to the A’ (tissue velocities) in (B). The calculated E/E’ratio was 8 in the normal and 15 in the diabetic subject.
Lipotoxicity and mitochondrial dysfunction
Insulin resistance results in a reduction of myocardial energy supply due to changes in substrate utilization from glucose to free fatty acids. It impairs myocardial glucose utilization and leads to excess fatty acid uptake into cardiomyocytes and eventually induces mitochondrial dysfunction, reduction in ATP availability and eventual cell death (lipotoxicity) [10]. The defect in myocardial energy production impairs myocyte contractile function, manifesting initially as diastolic function. Excess myocardial triglyceride content (myocardial steatosis) is demonstrable on proton-MR spectroscopy and has been correlated with echocardiographic left ventricular diastolic dysfunction as well as with longitudinal strain measurements [12].
Impaired coronary flow reserve in diabetes
Several factors: reduced NO production, AGEs- mediated stiffening of coronary media, and perivascular fibrosis, contribute to a reduction in coronary flow reserve. In addition, cardiomyocyte hypertrophy is associated with a reduction in capillary density (microvascular rarefaction) that leads to impaired myocardial perfusion, lowers NO bioavailability and contributes to myocardial stiffness that is typical of diastolic dysfunction. Tissue hypoxia results in the further release of ROS leading to myocyte cell death (apoptosis) and a decline in systolic function accompanied by remodeling with ventricular dilatation [11].
Microvascular disease and ischemia
Microangiopathy, characterized by thickening of the capillary basement membrane and the media of the arteriole, and associated with perivascular fibrosis, has been observed in autopsy samples of diabetic patients. Microaneurysms and spiral deformation of microvessels in the myocardium of type 2 DM, similar to retinal vascular changes of diabetes, have also been described. It has been shown that expression of vascular endothelial cell growth factor (VEGF) in the heart is downregulated in diabetes and that this downregulation is closely associated with the reduction in capillary density, apoptosis of endothelial cells and interstitial fibrosis [13].
The microangiopathy in diabetes explains why microalbuminuria/proteinuria is not only associated with nephropathy, but with widespread microvascular disease, including the heart. In the heart, diabetic autonomic neuropathy contributes to impaired autoregulation and lack of flow reserve, a factor that may account for increased rates of sudden cardiac death as well as a higher overall cardiovascular mortality rate in diabetic patients.
Autoimmunity
In type 1 DM subjects, cardiac myosin autoantibody and troponin T release are thought to trigger an immune response leading to myocyte cell death and replacement fibrosis. It has been proposed that this immune response, combined with the effects of worsening tissue hypoxia described above, probably provide the stimulus to the development of eccentric ventricular remodeling, resulting in ventricular dilatation and systolic dysfunction leading to the picture of dilated cardiomyopathy that has been described in type 1DM [11] (see below).
Development of myocardial dysfunction
As explained above, excess chronic oxidative stress produced by the release of ROS from the mitochondria and from proinflammatory cytokines and leucocytes, cause direct damage to plasma membrane cell organelles leading to myocardial damage. This may account for the increased oxidative injury resulting in the excessive morbidity and mortality after myocardial infarction in patients with diabetes when compared to patients without diabetes. Oxidative stress is the unifying factor in the development of diabetes-related cardiac complications, including atherosclerosis (Figure 1).
Myocardial damage in the absence of epicardial coronary disease (macrovascular) is most likely related to microvascular dysfunction, leading to diabetic cardiomyopathy (DMCMO).
Recent evidence has shown that insulin resistance-induced arterial stiffness in normotensive subjects contributes to diastolic dysfunction independent of age, blood pressure and body mass index. Together with activation of the sympathetic nervous system, the increase in afterload and impaired ventricular-vascular coupling as a result of arterial stiffness increase likelihood of heart failure development in these subjects [14].
Based on the fact that subjects with diastolic dysfunction have more LV hypertrophy and stiffness (attributed to AGEs deposition and stiff cardiomyocytes), while subjects with systolic dysfunction have a dilated left ventricle (following cardiomyocyte cell death and replacement fibrosis) [15], a new model of diabetic cardiomyopathy has been proposed. In a recent review Seferovic and Paulus [11] proposed that the deposition of AGEs in the myocardium and coronary microvascular rarefaction contribute to both diastolic and systolic dysfunction. Hyperglycemia, lipotoxicity, and hyperinsulinemia lead to myocyte hypertrophy, increased diastolic stiffness and the development of the restrictive/HFPEF phenotype, typical in obese type 2 DM patients (Figure 1). Autoimmunity with myocyte cell death leads to the dilated/HFREF phenotype that is more prevalent in type 1 DM patients. In this model, more selective involvement of endothelial cells in the coronary microvasculature drives the progression to diastolic dysfunction, while cardiomyocyte damage and ensuing myocyte loss trigger eccentric remodeling and decline in systolic function. (Table 1).
Epidemiology
Because of the structural and functional changes that occur in DMCMO, subjects develop functional changes early in the course of their disease. Diastolic dysfunction is the most frequent echocardiographic finding in both type 1 DM and type 2 DM patients and precedes the development of symptoms. The prevalence of asymptomatic diastolic abnormalities detected on TDI in a population-based study is high (23%) with over a third developing heart failure at 5 years [16]. In the early stages of type 1 DM, subclinical myocardial dysfunction is frequent, but clinical signs of heart failure are infrequent and developed in 3.7% of subjects over a 12-year follow-up period in one study [17]. Subjects who developed heart failure were older and had a longer duration of diabetes (35±9 years); they had higher blood pressure and a higher prevalence of albuminuria and retinopathy than those without heart failure. Diabetic patients with microvascular complications showed the strongest association with cardiomyopathy and this relationship paralleled the duration and severity of hyperglycemia [16]. These findings have been confirmed in a recent large case-controlled study from the Swedish national registry that showed a fourfold increase in the risk of heart failure in type 1 DM, especially in subjects with poor glycemic control and impaired renal function [18].
Clinical markers and implications for treatment
Asymptomatic diastolic dysfunction offers an opportunity for the primary prevention of heart failure in at-risk diabetic subjects if changes can be detected early and appropriate therapy instituted. Since there is little evidence to support the use of biomarkers in detecting the early stages of DMCMO, a strategy of screening asymptomatic diabetic subjects for impaired LV function with natriuretic peptides is not recommended. It is suggested that diabetic subjects at risk of developing DMCMO (such as those with atrial fibrillation, microalbuminuria/proteinuria, autonomic neuropathy, retinopathy, metabolic syndrome) should undergo non-invasive imaging [13] using tissue Doppler imaging and strain rate imaging as well as magnetic resonance spectroscopy, to enable early detection of DMCMO.
Subclinical changes of diastolic dysfunction has been demonstrated across the spectrum of IR and may present before the onset of diabetes, presenting an even larger disease burden for primary prevention of heart failure in diabetes [14,19]. There is evidence that glycemic control and lifestyle measures started earlier in the course of the IR spectrum (early diabetes, prediabetes and metabolic syndrome) could address the metabolic milieu before they have become established and may be beneficial. In this respect the Diabetes Control and Complications Trial (DCCT)/Epidemiology of Diabetes Interventions and Complications (EDIC) study has shown that intensive treatment of hyperglycemia, targeting glycated hemoglobin levels below 7%, when initiated early in patients with short duration of diabetes and low cardiovascular risk, results in a significant 42% reduction of cardiovascular events in the long term [20].
Conclusion
The diagnosis of diabetic cardiomyopathy may be inferred when myocardial disease in patients with diabetes cannot be attributed to any other known cardiovascular disease. Metabolic and microvascular mechanisms contribute to the pathogenesis. At a subcellular level in the mitochondrion, oxidative stress, coupled with loss of normal microvessels and remodeling of the extracellular matrix, lead to an inflammatory state with decline in cardiomyocyte contractile function. The disease course consists of a hidden subclinical period, during which endothelial dysfunction drives the cellular and matrix changes that result in diastolic stiffness, while autoimmune responses result in myocyte loss and decline in systolic function.
Subjects at risk of DMCMO are those with a long duration of poorly controlled diabetes, evidence of microvascular disease elsewhere, atrial fibrillation and those with markers of insulin resistance such as central obesity and metabolic syndrome.