
Background
Potassium is a soft, silvery-white highly reactive cation belonging to the alkali metal group family in the periodic table. It is the most abundant cation in the human body as a whole, and the most widespread ion in its intracellular compartments.
On average, a western diet contains from 80-100 mEq of potassium per day, and under normal physiologic conditions, 90% of it is absorbed passively, leaving only 9.0 mmol for fecal excretion. The 3500-4000 mmol kept stored in the body are disproportionate to the diurnal plasma potassium levels which are normally maintained in the range of 3.5-5.3 mmol/L through a tight homeostasis mechanisms with the lowest levels being at night and in the early morning hours and the highest peak level in the afternoon hours. [1]
Once absorbed into the blood stream, it becomes the role of the kidney to match potassium intake to potassium output; requiring several hours, during which time the “internal potassium balance” under the influence of insulin and catecholamines maintains temporary homeostasis by shifting the potassium between the intracellular and extracellular spaces. Stimulation of the alpha receptors impairs potassium entry into the cells, and stimulation of the beta receptors promotes it by activating the sodium potassium ATPase pump.
The sodium-potassium ATPase pump is the gate-keeper enzyme located in the sarcolemma. It helps to safeguard 98% of potassium (approximately 144.0 mmol) retained inside the cell. This ensures the preservation of the vital potential difference across the cell membranes needed for proper cell function, especially the excitable cells such as nerve cells and the cardiac muscle cells. [2,3,4]
Normal physiology and pathophysiology of potassium
After its rapid absorption, potassium helps orchestrate its own body levels through the release of insulin and aldosterone. Other inherent body stimuli also found to control potassium body levels include beta-2 adrenergic receptors, alkaline blood PH, and cellular anabolism.
Release of Insulin and Aldosterone: Ingested potassium rapidly enters circulation. On reaching the portal circulation, it stimulates the pancreas to release insulin. Concurrently, the circulating potassium reaching the juxtaglomerular cells results in the release of renin. Renin, on reaching the liver, is converted to angiotensin I. Angiotensin I travels to the lungs where it is converted into angiotensin II. Angiotensin II then completes its journey back to the kidneys through the circulating blood to stimulate the zona glomerulosa to secrete aldosterone.
Internal Potassium Balance: The insulin released post-prandially acts primarily on the skeletal muscles, activating two pathways, the AKT-dependent pathway responsible for the insertion of the glucose transporter GLUT4 and the APK pathway activating the cellular sodium potassium ATPase to shift the potassium into the intracellular space. Unlike the AKT-dependent pathway, the APK pathway is unimpaired by neither metabolic syndrome nor chronic kidney disease [4] (Figure 1).
Excretion: Potassium filtered by the renal glomeruli is passively reabsorbed in the proximal tubule and loop of Henle in proportion to the amount of sodium and water delivered. Normally only about 10% of the filtered load reach the distal nephron.
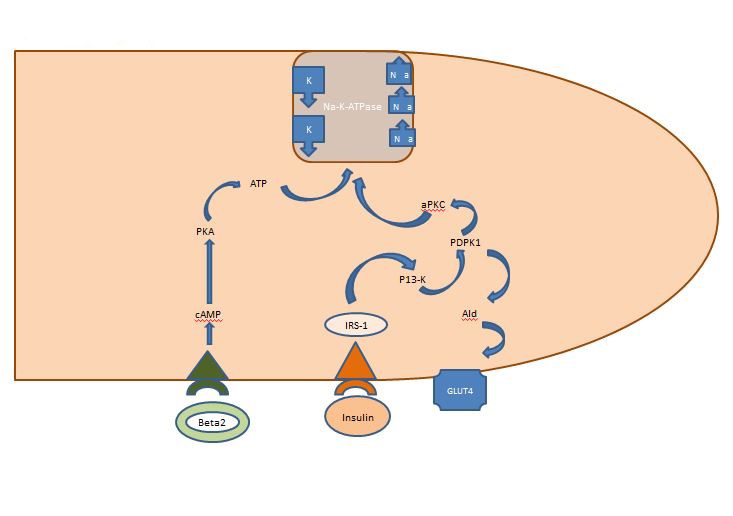
Figure 1. Action of insulin on a skeletal muscle cell. Insulin released post-prandially activates two pathways in skeletal muscles, the AKT-dependent pathway responsible for the insertion of the glucose transporter GLUT4 and the APK pathway activating the cellular sodium potassium ATPase to shift the potassium into the intracellular space.
At the beginning of the distal convoluted tubule, secretion of excess potassium commences and increases progressively as it advances further towards the distal nephron and into the collecting duct. This is mediated by the upregulation of hydrogen potassium ATPase on the alpha-intercalated cells [5].
The presence of higher potassium levels in the peritubular cells of the kidneys activates the RAAS system to release aldosterone, which activates the sodium potassium ATPase in the basolateral membrane, resulting in a decrease in the intracellular sodium which leads to the increased electrogenic transport of potassium uptake by hyperpolarizing the membrane voltage and allowing its excretion into urine [2].
In hyperkalemia, the quota of potassium excreted through the colon may increase by up to 30%, e.g., in cases of renal failure, where the potassium is then actively taken up by the activated sodium potassium ATPase pump in the colonic enterocytes’ basolateral membrane, to be excreted on the other side, into the colonic lumen through the apical large calcium-dependent potassium channels of the cells.
It is thus discernible from the above that the mechanism of potassium plasma level homeostasis is ordained mainly by the interaction of three simultaneous transactions - potassium intake, potassium intra/extracellular shifts and potassium urinary excretion, all of which ultimately rely on the sodium potassium pump.
To comprehend the mechanism of imminent danger from hyperkalemia and its management, one must understand the physiology of the action potential and the innards of the sodium potassium ATPase enzyme.
Electrophysiology of the action potential, i.e., ionic movement across the cell membranes, is determined by the difference in two potentials, a “chemical potential” in which the ions move down their concentration gradient and an “electrical potential” in which ions and molecules repel like charges, yielding the transmembrane potential (TMP), which is said to be +ve when the net movement of +ve ions is to the outside of the cell and vice versa.
Action potential of a non-pacemaker cardiomyocyte
There are five phases to an action potential, which begin and end at phase 4. The pumps involved in this process include the sarcolemma sodium calcium exchanger, calcium ATPase and, ultimately, the sodium potassium ATPase.
- Phase 4. The resting phase: this has a resting potential of -90 mV as a result of the constant outward movement of potassium via the inward rectifier channels. During this phase, both the sodium and calcium channels are closed.
- Phase 0. The depolarization phase: the firing of a pacemaker cell or its conduction through a neighboring cell triggers the rise of TMP to above -90 mV. At this point , the “fast sodium channels” start opening one by one, allowing sodium to enter into the cell, raising the TMP and, once enough fast sodium channels have opened to yield -70 mV, a self-sustaining inward sodium current is set into motion, rapidly depolarizing the TMP to 0 mV for a transient interim known as the "overshoot", at which point the time-dependent fast sodium channels close and the "long-opening” calcium channels open to raise the TMP to -40 mV and allow a small steady calcium influx down its concentration gradient.
- Phase 1. The early repolarization phase: this starts with the slightly +ve TMP and the brief opening of some potassium channels resulting in its flow to the outside of the cell, returning the TMP back to approximately 0 mV.
- Phase 2. The plateau phase: here the two counter currents are electrically balanced and result in the maintenance of the TMP balanced at just below 0 mV. “The long-opening calcium channels” are still open, resulting in a constant calcium flow into the cell. The delayed rectifier potassium channel allows the passage of potassium to the outside of the cell down its concentration gradient.
- Phase 3. The repolarization phase: during this phase, the calcium channels are gradually inactivated and the persistent flow of potassium to the outside of the cell thus exceeds the inward calcium flow, returning the potassium to the intracellular space and the sodium and calcium to the outside of the cell.
Action potential of a cardiac pacemaker cell
The cardiac pacemaker cells have an innate automaticity, allowing their depolarization in rhythmic cycles. The sinoatrial node (SAN) has the highest self-initiated depolarizing rhythm at a rate of 60-90/min, followed by the atrioventricular node (AVN) at a rate of 40-60/min and then the Purkinje fibers and ventricular muscle at 20-40/min.
The membrane potentials of pacemaker cells are unstable and their action potentials have no clear-cut phases. They have fewer inward rectifier potassium channels and their TMP never drops to below -60 mV, eliminating the role of the fast sodium channels that require a TMP of -90 mV resulting in the absence of the rapid depolarization phase.
At TMP >-60 mV, "funny/pacemaker" current is set into action with a spontaneous flow of ions through the slow sodium channels, depolarizing the TMP to <-50 mV and then back to -60 mV when the calcium channels close.
Current conduction
All the cardiomyocytes are electrically coupled through the gap junction, including the pacemaker cell. This facilitates the widespread depolarization of all neighboring cells, turning the heart into one functional unit in which the cell with the highest inherent rate becomes the "pacemaker".
Refractory period
The longer refractory period during the long plateau in phase 2 due to the slow calcium channels provides the time needed for the complete emptying of the ventricles before the next contraction. Refractory periods can be absolute (ARP), effective (ERP) or relative (RRP). In an ARP, the cell is absolutely unexcitable.
An ERP lasts from the ARP until the short segment of phase 3. A stimulus at this point could minimally depolarize the cell, but the level of depolarization is weaker than propagating an action potential to the neighboring cells.
RRP is brought about by an above normal stimulus, leading to the depolarization of the cell and the production of an action potential.
A “supra-normal period” is a hyperexcitable state during which a weaker than normal stimulus could lead to an arrhythmia, necessitating the synchronization during cardioversion to avoid ventricular fibrillation [6] (Figure 2).
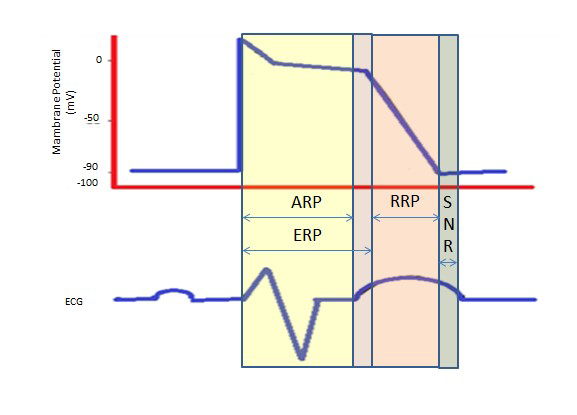
Figure 2. Refractory Periods. ARP: Absolute Refractory Period; ERP: Effective Refractory Period; RRP: Relative Refractory Period; SNR: Supranormal Refractory Period
Hyperkalemia, classification and causes
Classification
Hyperkalemia is classified as mild when levels are in the range of 5.5-6.0 mmol/L, moderate from 6.1-6.9 mmol/L and severe at levels of 7.0 mmol/L or greater, and at any level at which ECG changes occur [7].
Causes
Hyperkalemia occurs when compensatory mechanisms are no longer able to cope with the imbalance, which is why it is usually multifactorial.
- Increase in the intake of potassium via any route, e.g., dietary oral intake, or intravenous administration of potassium containing fluids like penicillin G.
- Retention by the kidneys: since potassium excretion depends on aldosterone and the delivery of a sufficient distal amount of sodium and water within the nephrons, conditions such as renal failure, adrenal insufficiency (Addison's disease) , hyporeninemic hypoaldosteronism type IV, renal tubular acidosis, especially in patients with diabetic nephropathy as well as any condition that promotes hypoperfusion as in volume depletion and congestive heart failure, will affect the intricate balance of potassium in the body and predispose to hyperkalemia.
- Adrenal insufficiency: this must be excluded in hyperkalemic patients, particularly in the presence of hyponatremia and muscle weakness. To screen for primary adrenal Insufficiency, a standard cosyntropin stimulation test is performed in which 0.25 mg synthetic cosyntropin is given as an intravenous bolus followed by plasma cortisol measurement 45 minutes to 1 hour later. Values less than 20 mcg/dL are suggestive of adrenal insufficiency.
- Drugs that retain potassium: prescription medication drugs which reduce sodium potassium ATPase activity such as beta-adrenergic receptor blockers, and drugs that reduce aldosterone secretion such as ACE and ARB inhibitors, non-steroidal anti-inflammatory drugs, and potassium-sparing diuretics, need close follow-up to avoid iatrogenic hyperkalemia, especially in the geriatric age group with their progressive decline in renal function as part of the aging process.
- Perturbations in the transcellular shift of potassium: this may occur with conditions of acidosis, hyperglycemia, hyperosmolality, severe exercise, tissue breakdown, hyperkalemic periodic paralysis, and with beta-adrenergic blockers. For every 0.1 unit decrease in blood PH, serum potassium increases by about 0.6 mmol/L (less if the acidosis is caused by organic acids) [2].
- Pseudo-hypoaldosteronism is a congenital autosomal recessive disease in which the kidneys are resistant to the actions of aldosterone.
- Pseudo-hyperkalemia must also not be overlooked: as the name implies, this occurs when there is elevated serum potassium in the presence of normal plasma potassium. It may be seen in hemolyzed blood, prolonged tight tourniquet during a blood sampling procedure, causing the extracellular release of potassium, with repeated clenching of the fist during phlebotomy, traumatic venepuncture, with leukocytosis and thrombocytosis, and in some uncommon genetic syndromes such as familial pseudo-hyperkalemia and hereditary spherocytosis. However, it could simply just be a result of a simple laboratory error.
Effects of hyperkalemia
Mild hyperkalemia is often asymptomatic, detected accidentally by laboratory tests, due to its vague symptoms such as malaise, muscle weakness and paraesthesia. Severe hyperkalemia will affect the neuromuscular function in the form of skeletal muscle weakness and paralysis; however, this is not a frequent presentation as the cardiac toxicity dominates the picture and is the preliminary presentation. Cardiac toxicity will usually present on the ECG in the following step-up escalating manner, although not necessarily so, depending on the etiology:
- At levels greater than 5.5 mEq/L, the increase in the conductance of potassium channels increases lkr current, leading to rapid repolarization in the form of a peaked T wave on the surface ECG. These T waves can be differentiated from those of myocardial infarction and CVA by their short duration ranging from 150-250 msec.
- At potassium levels greater than 6.5 mEq/L, a state of sustained subthreshold depolarization occurs, causing a delay in atrial and ventricular depolarization. The decrease in phase 0 of the action potential leads to a longer action potential, producing a delay in intraventricular and atrioventricular conduction. On the surface ECG, this will present with a flattening and loss of P waves and widening of QRS complexes. With increasing delay in the intraventricular conduction, the surface ECG starts to show signs of left and right bundle branch block. This can be differentiated from bundle branch disease by the fact that in hyperkalemia the delay persists throughout the QRS complex, not just during the initial or terminal portions, respectively.
- At 10 mEq/L, sinoatrial conduction no longer occurs and the accelerated junctional rhythm takes over. Ventricular arrhythmias develop with merging of the widened QRS complexes with the T waves eventually to form the classic sine-wave pattern. Once this occurs, VF and asystole are imminent and cardiac arrest will then ensue.
- Sometimes changes may be erratic and unpredictable and the ECG will jump from normal to asystole due to the variability in the etiological factors and their influential effects, e.g., rate of potassium change, calcium concentration, pH, and sodium concentration. Thus, hyperkalemia should be treated emergently whenever potassium levels become greater than 6.5 mmol/L, or in the presence of ECG manifestations of hyperkalemia regardless of the potassium level. Other reported associations with acute hyperkalemia include: picture of pseudo MI on the ECG recording, with massive ST-T segment as a result of derangements in myocyte repolarization, short PR and QT intervals, sinus tachycardia, sinus bradycardia, idioventricular rhythm, 1st and 2nd degree heart block [3].
Metabolic effects
Hyperkalemia leads to hyperchloremic metabolic acidosis as the hyperkalemia promotes the intracellular uptake of potassium in exchange for hydrogen ions. This creates intracellular alkalosis, suppressing kidney ammonia production in the proximal tubules, leading to a decrease in urinary ammonium and acid excretion and a type IV renal tubular acidosis [8].
Sodium potassium pump
The sodium potassium ATPase was discovered in 1957 by Skou, who was later awarded a share of the 1997 Nobel Prize in Chemistry for his discovery.
Skou was the first to discover the sodium potassium ATPase in the sarcolemma of the cardiac muscles' cell surface. Its presence was later detected in every eukaryotic single and multicellular organism.
The sodium potassium pump functions by linking the hydrolysis of ATP to the cellular export of three sodium ions in exchange for two potassium ions against their electrochemical gradients. It is the molecular target for digitalis and digoxin, which have been in use since the 18th century as foxglove extracts.
The action of the sodium potassium pump is regulated by a phosphoprotein phospholemman, whose unphosphorylation leads to the inhibition of the pump and whose phosphorylation leads to an increase in the pump activity. It has three phosphorylation sites, two palmitoylation sites and one glutathionylation site, which explains the multitude of signals capable of stimulating and inhibiting the pump.
The sodium potassium pump itself is an enzyme composed of multiple subunits with multiple isoforms. The presence of the alpha and beta subunits (mainly B1 in the heart) is essential for its function. Recently, a third protein gamma subunit has been identified in the kidneys, but to date its function remains unknown.
The alpha subunit is the catalytic core of the sodium potassium pump enzyme. It is approximately 100 kDa and contains the binding sites for sodium, potassium, ATP, and cardiotonic steroids such as ouabain. Only alpha 1 and alpha 2 display a significant presence in a normal cardiac myocyte and are functionally linked to the sodium calcium exchanger (NCX). Alpha 3 has been reported to replace alpha 2 in experimental heart failure models [2].
Data from recent experiments favor the involvement of both alpha 1 alpha 2 subunits of the pump in the regulation of the excitation-contraction (E-C) coupling. The alpha 1, which was found to be more evenly distributed across the sarcolemma, is thought to play more of a "housekeeping" role, controlling both contractility and the bulk intracellular sodium, while the alpha 2 whose expression is concentrated in the T-tubules along with other key components of E-C coupling is thought to focus mainly on contractility [2,9].
Known factors that can control the sodium potassium pump include: ATP, intracellular sodium, sub-sarcolemmal barriers and fuzzy spaces, membrane potential, intracellular signaling pathways (adrenergic signaling pathways, protein kinase A & C, nitric oxide, phospholemman), direct regulation by small molecules (lipids, endogenous cardiotonic steroids), other associated proteins (caveolae and caveolins, and ankyrin).
Conclusion
Hyperkalemia is a clinical challenge and may present in up to 10% of hospitalized patients [10]. Its end result is life-threatening. As all of the cells in the body are ultimately affected by the sodium potassium pump, and ischemic cardiac muscles are known to extrude their potassium extracellularly leading to a reduction in the arrhythmia threshold with the possibility of ventricular arrhythmias that aggravate the hypopolarization and lower the threshold even more, more studies need to be focused on the manipulation of the sodium potassium enzyme, as its control could favorably alter the outcomes of cardiac arrests and rewrite the current CPR guidelines.