

Introduction
Fabry disease (FD) (Online Mendelian Inheritance in Man [OMIM] number 301500) is an X-linked disease of the lysosomal metabolism resulting in a deficiency of the α-galactosidase A (GLA) enzyme [1]. The disease is characterised by accumulation of glycosphingolipids in different cell types. This process is responsible for a heterogenic phenotype with involvement of the skin, nervous system, kidneys and heart. Eventually, it can lead to death, mostly from cardiac, renal or cerebrovascular complications [1].
Epidemiology
FD is a rare disease. The prevalence in the general population is estimated to be around 0.02 to 0.09 per 10,000 [2]. The prevalence of the disease has been described as being 0.5-1% in patients with unexplained left ventricular hypertrophy [3]. In patients with end-stage renal disease on haemodialysis treatment, the prevalence of FD ranges between 0.2 and 1.2% [1]. In male patients with cryptogenic stroke the prevalence may be as high as 3-4% [1]. However, not all patients, who could be detected through screening, will show systemic signs of FD, therefore, the true prevalence may be underestimated [3].
Pathophysiology
FD is genetically caused by mutations of the GLA gene [1]. Hundreds of mutations within the GLA gene have been identified. These consist mostly of point mutations, such as missense or nonsense mutations, but small and large deletions are seen as well. Loss-of-function mutations are associated with classic forms of the disease, whereas other mutations may lead to a milder phenotype or late-onset variants [4]. The GLA gene is located on the X-chromosome, therefore the disease shows an X-linked inheritance pattern without any male-to-male transmission. However, it has been shown that even heterozygous women frequently have signs of significant multisystemic disease, though often present at a later age than their male counterparts [5].
The mutations in the GLA gene lead to a deficiency of α-galactosidase A, which again is responsible for the storage of neutral glycosphingolipids, notably globotriaosylceramide and galactosylceramide, in different tissue and cell types. This leads to cellular dysfunction, which consecutively induces inflammation, fibrosis, or both. This will eventually lead to organ dysfunction through at least some degree of irreversible tissue damage caused by perfusion deficiency [1].
In the heart, globotriaosylceramide can be accumulated in all cellular components, including cardiomyocytes, conduction system cells, valvular fibroblasts, endothelial cells, and vascular smooth muscle cells [1].
Clinical presentation
The clinical presentation can be highly heterogenous; however, classic FD is typically a slowly progressive disease with changing signs and symptoms over the course of time. Acroparaesthesiae, pain crises, angiokeratomas, Raynaud phenomenon, cornea verticillata, hearing impairment, dyshidrosis, bowel disturbances and lethargy can appear in childhood and adolescence. In early adulthood, with the progression of the disease, patients typically develop more extensive angiokeratoma, proteinuria, lipiduria or haematuria, oedema, fever, dyshidrosis, lymphoedema, heat sensitivity, diarrhoea or abdominal pain. Patients develop clinically detectable heart disease, impaired renal function and strokes, typically in adulthood; however, cases with cardiac involvement in childhood have been described [6-8].
Cardiac involvement
Cardiac manifestations include valvular disease, left and right ventricular hypertrophy, ischaemia, electrocardiogram (ECG) abnormalities and arrhythmias [1]. Left-sided heart valves tend to be thickened and distorted, and usually show regurgitation, though they will rarely require valvular surgery [9].
Before the enzyme replacement therapy (ERT) era, almost all patients with FD progressed to left ventricular hypertrophy (LVH), despite the exclusion of patients with arterial hypertension, with a mean age at onset of about 44 years for men and 55 years for women [10]. Hypertrophy may begin as concentric remodelling and may later proceed to concentric hypertrophy, typically involving the septum and posterior wall [1]. Myocardial infarction is rare in patients with FD; however, patients commonly complain of angina or chest pain, which is most likely caused by increased myocardial oxygen demand, small vessel disease and endothelial dysfunction leading to microvascular angina [9]. However, this does not exclude coronary artery disease and additional cardiovascular risk factors should be avoided or treated. ECG abnormalities may include voltage criteria for LVH and repolarisation changes, short PQ-interval, bundle branch block, atrioventricular conduction delay, and progressive sinus node dysfunction [1]. Arrhythmias are commonly named as the cause of sudden cardiac death in patients with FD. However, a small retrospective study showed that patients with FD and implantable cardioverter defibrillators (ICDs) had a high need for antibradycardia pacing, but ventricular tachycardia was only rarely detected [11]. There appears to be no genetic predictor for the cardiac outcome [12].
Diagnosis
Since the availability of ERT, screening for FD has become more meaningful as effective treatment is available for these patients. With the increasing number of patients screened, the rate of patients with borderline signs of FD will also increase, leading to uncertain diagnoses.
The Heart Failure Society of America Practice Guidelines recommend genetic screening for FD in all men with sporadic or non-autosomal dominant (without male-to-male) transmission of unexplained cardiac hypertrophy [13]. Certain clinical signs may serve as red flags to help select patients for screening, such as a PQ-interval of <120 ms, sinus bradycardia, hypertrophied papillary muscle on echocardiogram or myocardial late enhancement of the inferoposterolateral region on cardiac magnetic resonance imaging. However, these signs are not fully specific for FD. Low voltage on ECG and severe LVH in patients younger than 20 years is unlikely to be consistent with FD [14].
For a definite diagnosis of FD, patients need to carry a GLA mutation. Male patients additionally need a lysosomal α-galactosidase A deficiency of ≤5% of the mean reference value in leukocytes. Moreover, both sexes need to show characteristic signs of FD, increased plasma (lyso) Gb3 or a family member with a definite FD diagnosis (Figure 1). However, the diagnosis remains uncertain if the patient presents with only a non-specific FD sign such as LVH, stroke at a young age, or proteinuria, and does not fulfil the criteria for a definite diagnosis, but has a GLA genetic variance of unknown significance (GVUS), in which case the diagnostic algorithm depicted in Figure 2 can be applied [14].
Figure 1. Definition of a definite diagnosis of Fabry disease as suggested by Smid et al [14]
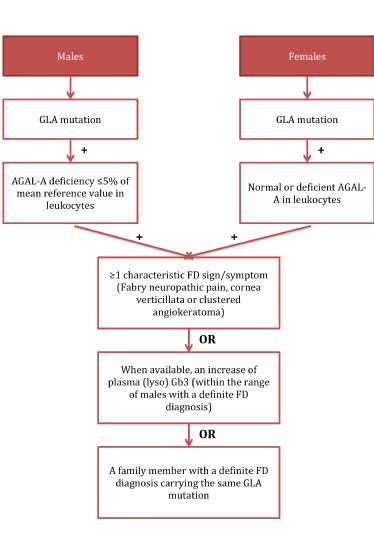
FD: Fabry disease; AGAL-A: lysosomal a-galactosidase A enzyme; GLA: a-galactosidase A gene; GLA mutation: defined as any abnormality found in GLA gene; Lyso Gb3: globotriaosylsphingosine
Figure 2. Diagnostic algorithm for subjects presenting with isolated LVH and an uncertain diagnosis of Anderson-Fabry disease as proposed by Smid et al [14].
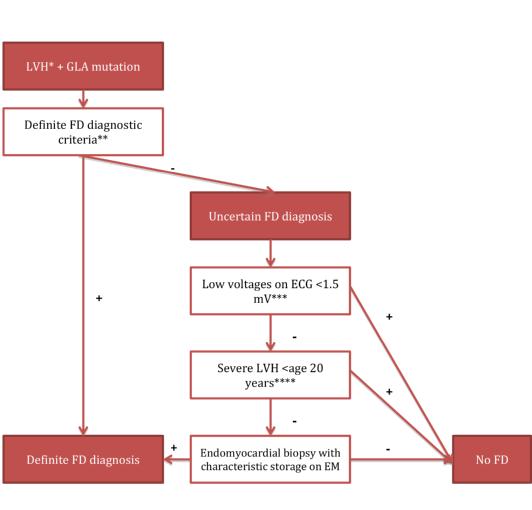
*LVH: left ventricular hypertrophy defined as an MWTd >12 mm; ** see text above; *** low voltages on ECG defined as the total sum of the amplitude of the QRS complex in I, II, III >1.5 mV;
**** severe LVH was defined as MWT >15 mm; FD: Fabry disease; EM: electron microscopy; GLA: α-galactosidase A gene
Conclusion
Fabry disease is a rare genetic disease with multisystemic consequences. Though showing an X-linked inheritance pattern, phenotypical expression may not be typical for X-linked diseases, as women usually also show signs of the disease. Patients can present with a variety of symptoms from the cardiovascular, cerebrovascular, renal or dermatological system. The disease is progressive and can lead to death.