

BackgRound
Definition
Hypertrophic cardiomyopathy (HCM) is defined as unexplained left ventricular hypertrophy (LVH) in the absence of other cardiac or systemic conditions known to produce comparable ventricular wall thickness (1–3). Some cases are not genotypically affected but the majority are. Hypertrophic cardiomyopathy is the prototype of ventricular hypertrophy of genetic origin and occurs in 1/500 in the general population (4).
Diagnosis
Diagnosis is mainly established by non invasive cardiac imaging - transthoracic echocardiography (TTE) and cardiac magnetic resonance. These typically will uncover asymmetric cardiac hypertrophy with diastolic dysfunction which can be associated with outflow tract obstruction and mitral regurgitation due to an abnormal anatomy of the mitral apparatus. European guidelines recommend a wall thickness ≥ 15 mm for adults and a Z-score >2 for children to retain the diagnosis regardless of the tool used for measurement.
Advances
Hypertrophic cardiomyopathy has remained idiopathic for more than one century with a well documented autosomal dominant mode of inheritance. Since the 1990s, several studies have rendered the identification of over 1,500 mutations (5) located in more than 11 genes possible. Myocyte disarray, fibrosis and small vessel disease are its main histological features (6).
The cardiac sarcomere
Most mutations occur in gene encoding components of the cardiac sarcomere, which is the fundamental contractile unit of the cardiac muscle. Microscopically, the sarcomere is limited by the Z lines to which the thin filaments are anchored (Figure 1). Rarely, mutations occur in components of the Z-disc or calcium-handling proteins. Hypertrophy may also be associated with specific syndromes that mimic HCM but that do not affect the sarcomere. Accordingly, cardiac hypertrophy may be classified according to whether the sarcomere is involved or not. Sarcomeric mutations include components of the thick myosin filament (MYH7, MYL2, MYL3…), the intermediate filament (MYBPC3), the thin actin filament (TNNT2, TNNI3, TNNC1, TPM, ACTC1), the Z-disc (ACTN2, MYOZ2) and some calcium handling proteins (junctophillin, phospholamban). All other mutations are non sarcomeric.
Mutations
The majority of mutations occur as a replacement of an amino acid by another (missense mutations). These replacements alter the physical and functional properties of proteins incorporated in the sarcomere which may trigger hypertrophic signals. More rarely, insertions or deletions of nucleotides occur and lead to frameshift mutations which heavily alter the mRNA translation and the proteins properties. In all, the majority of mutations are “private”, i.e specific to a single family (7). Molecular mechanisms leading to hypertrophy are poorly understood, nevertheless, some pathways are more likely to underlie the disease; they associate a disturbed biomechanical stress, impaired calcium cycling and sensitivity, altered energy homeostasis and increased fibrosis (8).
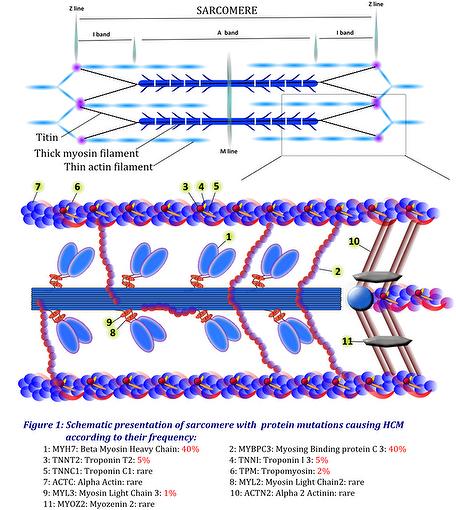
I - Testing
Genetic testing, initially performed for research purposes, has become available in daily practice thanks to several commercial tests (Genedx, Correlagen, transgenomic-Familion..) which cost between 3,000 and 5,000 Euros (https://www.genetests.org/). The identification of a wide range of mutations whose pathogenicity may be difficult to demonstrate has since occurred (9). It has been able to establish pathogenecity in 11 genes and two group mutations.
A) Established pathogenecity
Pathogenicity is well established in 11 genes, among which mutations in the MYH7 and MYBPC3 genes account for about 80% of established mutations (Table 1).
Some rare mutations with a lower evidence for pathogenicity have also been identified (MYH6: α myosin heavy chain; TTN: Titin; CASQ2: Calsequestrin….)
Table 1: Genes with established pathogenicity for HCM (adapted from HGNC and OMIM database)
|
Gene |
% of established mutations |
Location |
Name (HGNC) |
Phenotype |
OMIM |
Muscular component |
|---|---|---|---|---|---|---|
|
MYH7 |
40 |
14q11.2 |
Myosin, heavy chain, cardiac muscle, Beta |
CMH1 (192600) |
160760 |
Sarcomere, thick filament |
|
MYBPC3 |
40 |
11p11.2 |
Myosin binding protein, cardiac |
CMH4 (115197) |
600958 |
Sarcomere, intermediate filament |
|
TNNT2 |
5 |
1q32.1 |
Troponin T type 2 (cardiac) |
CMH2 (115195) |
191045 |
Sarcomere, thin filament |
|
TNNI3 |
5 |
19q13.42 |
Troponin I, type 3 |
CMH3 (613690) |
191044 |
Sarcomere, thin filament |
|
TPM1 |
2 |
15q22.2 |
Tropomyosin 1 (α) |
CMH 3 (115196) |
191010 |
Sarcomere, thin filament |
|
MYL2 |
? |
12q24.11 |
Myosin, light chain 2, regulatory, cardiac, slow |
CMH 10 (608758) |
160781 |
Sarcomere, thick filament |
|
MYL3 |
1 |
3p21.31 |
Myosin, light chain 3, alkali, ventricular, skeletal slow |
CMH 8 (608751) |
160790 |
Sarcomere, thick filament |
|
ACTC1 |
? |
15q14 |
Actin, alpha, cardiac muscle 1 |
CMH 11 (612098) |
102540 |
Sarcomere, thin filament |
|
ACTN2 |
? |
1q43 |
Actinin, α2 |
612158 |
102573 |
Z-disc |
|
TNNC1 |
? |
3p21.1 |
Troponin C type1 (slow) |
CMH 8 (613243) |
191040 |
Sarcomere, thin filament |
|
MYOZ2 |
? |
4q26 |
Myozenin 2 |
CMH 16 (613838) |
605602 |
Z-disc |
B) Phenotype diversity
Phenotype diversity in HCM is the main result of its genetic diversity. Nevertheless, certain mutations together cause the bulk of the more common clinical features. They are the following mutations:
MYH7 seem to produce broad phenotypes with high penetrance.
MYBPC3 seem to be associated with a later onset of a mild hypertrophy and a good prognosis.
TNNT2, despite mild hypertrophy, seem to cause SCD more frequently.
MYH7. In addition to hypertrophy, may lead to dilated cardiomyopathy (DCM), left ventricular non compaction (LVNC), Laing distal myopathy and myopathic type scapuloperoneal syndrome (10).
TNNT2 cause DCM, restrictive cardiomyopathy (RCM) and LVNC (11).
MYBPC3 seem to exclusively cause HCM.
Phenotypes orienting to certain genes as described above may improve the pretest probability by orienting the clinician to some potential mutations and thus improving the probability of getting a positive result.
C) Group mutations and clinical features
In addition to accurately identifying patients with HCM, genetic testing has allowed two major groups to be defined:
The first group is referred to as the genotype positive/phenotype negative (G+/P-) group. It includes mutation carriers without overt hypertrophy.
The second group includes patients with ventricular hypertrophy caused by non sarcomeric mutations and it is referred to as phenocopies.
- G+/P- group: This group is important as it may be the target for therapeutics aiming to stop and even to prevent the disease. This group has not been included in SCD risk stratification studies. Cardiac events are thought to be uncommon in this group. Therefore, the decision for ICD implantation before overt hypertrophy may be critical. This group requires a thorough and regular follow up as patients can develop hypertrophy at any time, mainly during adolescence. Some echocardiographic signs may precede the development of overt hypertrophy; they include early diastolic dysfunction, myocardial crypts and scarring and elongated mitral leaflets. Cardiac magnetic resonance is a valuable tool for a better analysis.
- The phenocopies group presents with various clinical, electrocardiographic and echocardiographic signs which are well summarised in the guidelines (12). When present, these signs should raise the possibility of HCM syndromes and lead to specific diagnostic tests. This is a complex group that may lead to cardiac hypertrophy by various mechanisms (13). These may be summarised as glycogen storage disease [GAA mutations (Pompe’s disease), PRKAG2 mutations…], lysosomal storage disease [GLA mutations (Fabry disease), LAMP2 mutations (Danon disease) …], mitochondrial disorders, cardiocutaneous syndromes or RASopathies, neuromuscular disorders, lipodystrophic syndromes, amyloidosis… (14). The presence of preexcitation at the ECG is a key feature orienting storage disease. Among those syndromes, PRKAG2, GLA and LAMP2 mutations are included in genetic test panels (Table 2).
Table 2: Clinical features associated to PRKAG2, GLA and LAMP2 mutations.
|
Gene mutation |
Protein mutation |
Inheritance |
Gene |
Clinical features |
|---|---|---|---|---|
|
PRKAG2 |
Gamma subunit of AMP-dependant protein kinase 2 |
Autosomic recessive |
7q36.1 |
Hypotonia; failure to thrive Hypoglycemia; Hepatomegaly; growth retardation |
|
GLA |
Alpha galactosidase |
X linked |
Xq22.1 |
Fatigue; acroparesthesia; proteinuria; renal failure, corneal opacity, anhidrosis; angiokeratoma; neuropathy |
|
LAMP2 |
Lysosome associated membrane protein 2 |
X linked |
Xq24 |
Proximal myopathy; raised CPK; cognitive impairment, visual impairment; WPW |
Legend: AMP: Adenosine monophosphate, WPW: Wolff Parkinson White
D) Genetic markers in evaluation of risk
Identifying causative genes of HCM was thought to improve risk stratification and gene mutations were initially designated as “malignant” or “benign” (15,16). Soon thereafter though, this classification proved limited as it not reproducible.
It is now admitted that single mutations do not accurately predict prognosis.
Nevertheless, double mutation carriers may have a higher risk of SCD and they are included in the ACC/AHA guidelines as modulators whom may support the decision for ICD implantation in some cases. Genetic markers nevertheless are not included in the recent SCD risk stratification models (17).
II) Benefits and limitations
Confirming diagnosis
Genetic screening is a valuable tool that may help to confirm the diagnosis of HCM even in ambiguous situations. This type of screening also helps to identify high risk patients before the occurrence of overt hypertrophy and removing the uncertainty for those with a negative test especially in related familial screening. As HCM may affect 50% of offspring, a prenatal and even a preimplantation test may be helpful. Genetic analysis is also the best way to better understand this complex disease where progress is still being made.
Predictive value
Tests are positive for 2/3 of patients with a history of HCM and 1/3 of those with first diagnosed LVH (18). Reliable results are better provided by experienced and certified centers. To improve the results, genetic screening should be performed in most clinically symptomatic patients. Therefore a) in the absence of a known mutation, a negative testing does not rule out HCM; b) patients with unexplained ventricular hypertrophy and a negative genetic test may be HCM carriers and should be closely followed; and c) when negative, genetic testing won’t be helpful for familial screening and clinical follow-up is therefore more appropriate.
Clinical and prognostic contribution
Positive testing doesn’t accurately predict phenotype or prognosis. This is mainly the result of the genetic variability which induces wide ranges of mutations seem to be at a higher risk for SCD. As a consequence a) prognosis assessing should be based on the common clinical parameters and the SCD risk stratification model and b) risk assessment in patients G+/P- may be critical as, to our best knowledge, this group was not included in studies dealing with prognosis in HCM.
Interpretation
Several mutations are of “unknown significance”. Those are unclassified mutations whose association with the disease risk is unknown. For instance, these variants are not useful for familial screening and require continuous reevaluation.
Table 3: Benefits and limitations of genetic testing
| Benefits | Limitations |
|---|---|
| Confirm HCM even in ambiguous cases and before overt hypertrophy | Limited positive predictive value |
| Rule out non affected cases | In the absence of a known mutation, a negative test doesn’t rule out HCM |
| Prenatal and preimplantation diagnosis | Limited clinical and prognostic contribution |
| Test interpretation may be challenging |
III - Automated DNA sequencing
Genetic screening has become feasible in daily practice with the introduction of automated DNA sequencing which allows accurate and reproducible diagnosis. Laboratories provide panels including causative genes, phenocopies and other genes possibly associated with HCM.
Figure 2: Results of genetic screening in clinical practice
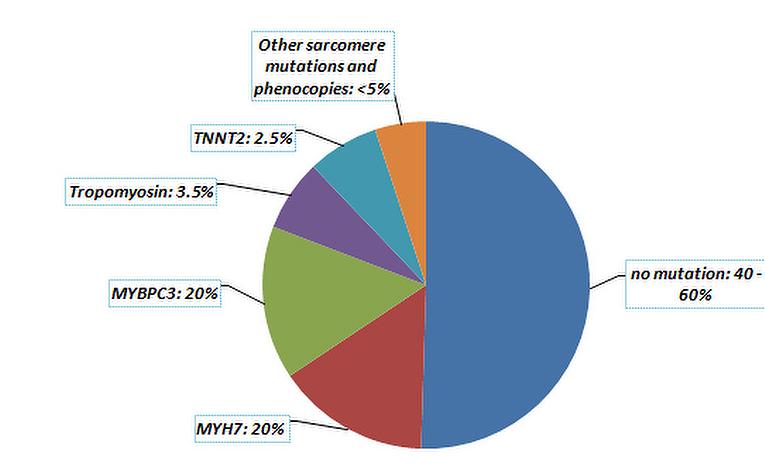
Genetic heterogeneity is so great that the pathogenicity of some identified mutations during screening can’t be demonstrated. However, to be considered pathogenic, a mutation should fulfill the following criteria: (19)
- Cosegregates with the HCM phenotype in family members
- Previously reported or identified as a cause of HCM
- Absent from unrelated and ethnic-matched normal controls
- Protein structure and function is importantly altered
- Amino acid sequence change in a region of the protein otherwise highly conserved through evolution with virtually no variation observed among species, suggesting its importance to basic cellular function
If the mutation does not fulfill the above criteria, it is classified as: variant of unknown significance (VUS) or non-pathogenic variant.
It should be noted that these tests should be regularly updated as several mutations may be reclassified (20). Genetic databases such as the ESP database, 1,000 Genomes project and clinvar together contribute to the sorting of several identified gene mutations.
IV - Common scenarios reviewed
Facing some confusion due to genetic testing, clinicians should keep in mind that this exam is meant to help clinical decision-making but not replace it. Some common clinical scenarios may present to clinicians and here is how to address some common clinical scenarios (Figure 3).
- Patients presenting with highly probable LVH of non-genetic origin (hypertension, aortic stenosis…) don’t require genetic testing unless some criteria expose ambiguity (familial SCD..) or ruling out HCM seems mandatory (athletes).
- If HCM is highly probable, genetic testing will accurately confirm the diagnosis which will facilitate familial screening and identify affected members even before overt hypertrophy.
- In the case of negative testing or identification of a VUS, a patient should still be considered as an HCM carrier and treated as such. Familial screening will only be based on clinical exam and TTE. It remains possible to repeat the tests to follow the advances as they are made in the field of genetic testing.
- Some clinical features, uncommon in HCM are of great importance in orienting to diagnosis of phenocopies such as preexcitation, myopathic syndrome, hepatomegaly etc.
Figure 3: Practical approach in suspicion of HCM
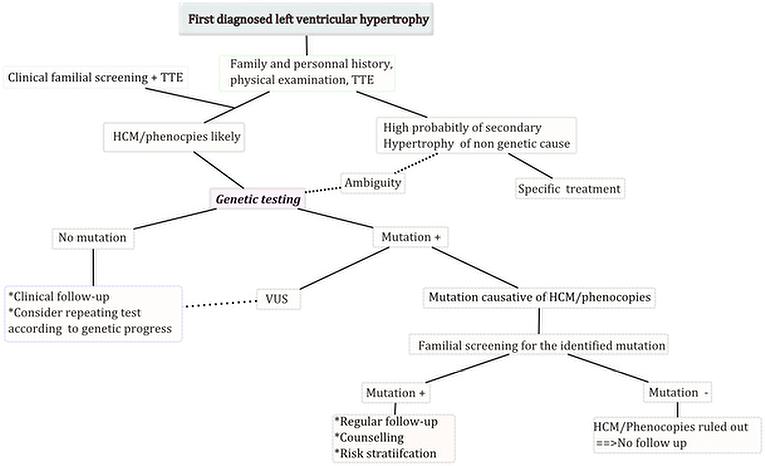
Legend: TTE: transthoracic echocardiography, VUS: variant of unknown significance
Conclusions
For a thorough management of patients with HCM, genetic testing is available and invaluable. Major progress has been made in with the discovery of several mutations that have uncovered marked genotypic and phenotypic heterogeneity of the disease. These tests are to be performed in certified diagnostic laboratories. The best indication is for testing patients that have fulfilled the diagnostic criteria for HCM allowing further familial screening. Some other potential benefits are offered to clinicians like prenatal and preimplantation testing; confirming or infirming the diagnosis in ambiguous situations and allowing a better understanding of the disease with further progress; however we should also be aware of the limitations of such testing.