
Background
Although dilated cardiomyopathy (DCM) is far less common than coronary artery disease and arterial hypertension, DCM is the third cause of heart failure (HF). It typically affects young adults. Numerous conditions and diseases leading to DCM can be treated effectively however the diagnostic pathway is difficult and requires comprehensive knowledge as well as access to sophisticated diagnostic methods, such as cardiac magnetic resonance, endomyocardial biopsy and genetic testing. To structure and simplify DCM patient flow, a three-staged diagnostic pathway is proposed.
I - Presentation
Definition - Cardiomyopathy is “a myocardial disorder in which the heart muscle is structurally and functionally abnormal, in the absence of coronary artery disease, hypertension, valvular heart disease and congenital heart disease sufficient to cause the observed myocardial abnormality” (1). Thus, an initial and comprehensive exclusion of those four causes of heart damage and failure which are coronary artery disease, hypertension, valvular heart disease and congenital heart disease should be performed.
Types - Dilated (DCM), hypertrophic (HCM) restrictive (RCM), and arrhythmogenic right ventricular (ARVC) are the four structurally and functionally distinct clinically-orientated classes of cardiomyopathies (2). However, types may, as end-stage (dilated) HCM, or ARVC with predominantly left ventricular involvement often overlap each other or mimic one other.
Cardiomyopathies are further classified into familial and non-familial forms.
Dilated cardiomyopathy - Regardless of the cause of the disease, dilated cardiomyopathy is best described as a progressive ventricular wall thinning and dilatation accompanied with gradual functional impairment (3). The phenotype of DCM is established by means of imaging studies – echocardiography being the most common (4).
Echocardiographic criteria: Echocardiography is used to establish the type of cardiomyopathy. The cardiomyopathy is considered dilated if the following criteria can be observed 1) left ventricular end-diastolic diameter (LVEDd) > 117% of the age and body surface 2) left ventricular systolic dysfunction (LVSD) defined by left ventricular ejection fraction (LVEF) < 45% and/or 3) fractional shortening (FS) < 25%.
In a majority of patients, dilated cardiomyopathy involves the inflammatory process and genetics however the causes are usually largely unknown.
Epidemiology: The most reliable data on the epidemiology of DCM comes from a study conducted between 1975 and 1984 in Olmsted County, Minnesota. It estimated DCM prevalence at 35.5/100,000 inhabitants (5). The annual incidence of DCM is approximately 5 to 8 per 100,000. DCM is a major cause of sudden death in young adults (6) and is currently the most frequent diagnosis in patients referred for heart transplantation.
Prognosis: Dilated cardiomyopathy is a severe myocardial disease that results in high morbidity and mortality, especially in young adults. The course of the disease is unpredictable, from relatively mild to severe and rapid, progressing to death. Typically, the prognosis is poor with a 5-year mortality of 46%.
II – Forms
A) Etiology
The etiology of DCM is heterogeneous. In developed countries CAD and myocardial infarction (MI) are the most common causes of HF, approximating 50-75% of all HF patients. The terms “non-ischemic cardiomyopathy” have been used to describe dilated cardiomyopathy however they are no longer recommended. Potentially reversible myocardial ischemia must always be sought actively, as effective therapy (medication and revascularisation) can favorably alter outcome. The presence of CAD and MI of course exclude the diagnosis of cardiomyopathy. Despite careful medical history and non-invasive examinations, the causative role of CAD in the development of HF has been shown only after coronary angiography in up to 7 % of patients with initially unexplained DCM (7).
The phenotype of DCM is common for numerous heart conditions. Therefore, the etiology is frequently difficult to identify. Generally, the inflammatory process, inherited forms and idiopathic DCM are equally responsible for more than 90% of cases. Other diagnosable causes of DCM, which always should be considered in the diagnostic process, include: remaining types of cardiomyopathies, which may either mimic or progress to DCM, connective tissue diseases, endocrinologic disorders, infiltrative diseases, medications and toxins, tachycardia-induced DCM and lastly miscellaneous causes. Most of these various diseases can be identified after careful medical history, basic laboratory and imaging studies. However, some disorders, especially infiltrative diseases may require more sophisticated examinations, such as laboratory tests, cardiac magnetic resonance (CMR), or the histopathopatological studies (8).
Once these causes of DCM have been excluded, the etiology of DCM can be either genetic (familial) or non-genetic. Further diagnostic steps are based on endomyocardial biopsy and state-of-the-art bioptates assessment as well as genetic studies (37). The genetic nature of DCM is increasingly recognised. However, at this stage, only 35% of DCM patients have confirmed causative mutations (9). In the non-genetic causes, persistent myocardial inflammation as a consequence of myocarditis is frequent.
B) Familial (genetic)
Idiopathic DCM is considered familial (genetic) when more than one first-degree relative has been diagnosed with DCM. It is estimated that familial DCM can be diagnosed in 20 to over 50% of patients with an initial diagnosis of IDC (10). However, the true frequency of familial DCM is probably underestimated because some of the initially healthy relatives may develop signs, mostly echocardiographic, and symptoms of the disease during follow-up examinations. Apart from positive three to four generation family history and detailed echocardiographic examination of asymptomatic relatives, the phenotype of familial DCM is otherwise indistinguishable from other forms of DCM. Therefore, confirmation of the causative mutation from detailed genetic analysis and positive family history are necessary to establish the diagnosis (9). The great majority of familial DCM is transmitted in an autosomal dominance inheritance pattern; however, other modes of inheritance such as autosomal recessive, X-linked, and mitochondrial have also been described (10). So far, numerous mutations in over 30 genes have been identified. The spectrum of genes associated with DCM is broad. Unlike hypertrophic cardiomyopathy which is primarily a disease of sarcomeric proteins, the genetic and molecular basis of DCM is more heterogeneous (11). On the molecular level, a wide variety of defected proteins, such as proteins comprising of a nuclear envelope, the cardiac sarcomere, ion channels, transcription factors, and the dystrophin-associated cytoskeletal complex have been implicated in the pathology of DCM (12, 13). The other unresolved issue is the presence of mutations in typical familial DCM genes in patients who have neither family history nor echocardiographic abnormalities.
The role of genetic testing in cardiomyopathies has previously been underlined by the American and European Societies. However, the most detailed recommendations on the subject come from the 2009 Guidelines of the American Heart Failure Society (14). This document provides detailed recommendations for obtaining a family history, screening family members, genetic counseling, genetic testing, and treatment. Genetic testing for HCM and ARVC is well validated. Nevertheless, genetic evaluation in DCM is also recommended. The other important issue, addressed by the guidelines, is the genetic overlap between cardiomyopathies. Probably one of the most striking examples of genetic overlap is DCM and HCM. Whereas in DCM, mutations of sarcomeric proteins cause severe contractile dysfunction, different mutations in the same sarcomeric genes lead to the opposite functional changes that are typically seen in HCM.
C) Non familial (non genetic)
The search for etiology of sporadic cardiomyopathy is even more challenging than familial DCM. Accumulating evidence, from basic and clinical studies, has paved the way to the concept of inflammation and/or autoimmunisation as the primary cause(s) of otherwise unexplained cardiomyopathy. Source studies by Felker at al. and from the Myocarditis Treatment Trial have consistently reported myocarditis as the cause of DCM in 9% and 10% of patients, respectively. Nevertheless, in half of the studied patients with DCM, etiology remained undefined (15). Dilated cardiomyopathy due to inflammation and sub-sequent autoimmunisation is probably more common than was previously thought. Novel imaging software such as CMR with tissue differentiation and techniques of molecular biology used in the assessment of cardiac bioptates, can detect inflammatory processes in tissues which would had been previously described as normal (16).
D) Inflammatory
Inflammatory dilated cardiomyopathy (DCMI) is a late and serious consequence of the complex interplay of the infectious agent, most often a virus, and the (auto)-immunologic response, which primarily develops in susceptible individuals (according to a genetic factor) (17). Although the DCMI phenotype is indistinguishable from the typical DCM, the diagnosis of DCMI cannot be made without endomyocardial biopsy (EMB). The state-of-the-art assessment of EMB specimens should encompass not only histo-pathological examination (Dallas criteria) but more importantly immuno-histochemistry and molecular biology techniques to detect DNA or RNA of infectious agent (18- 20). Rarely, myocardial inflammation can be caused by a variety of non-viral infectious organisms including bacteria, fungi, or protozoal agents as well as non-infectious diseases such as systemic diseases, (auto) immune disorders, hypersensitivity reactions, and toxins.
Histological diagnosis of myocarditis (Dallas criteria):
- Active myocarditis – is defined as “an inflammatory infiltrate of the myocardium with necrosis and/or degeneration of adjacent myocytes not typical of the ischemic damage associated with CAD”. In most cases infiltrates are mononuclear, but may be neutrophilic, or eosinophilic (in rare cases of eosinophilic myocarditis).
- Borderline myocarditis – is diagnosed when the inflammatory infiltrate is too sparse, or myocyte injury is not demonstrated.
Immunohistochemical diagnosis and quantification of inflammatory process (21):
- Abnormal findings – more than 2 CD3+ T-lymphocytes per high-power filed (HPF) (400-fold magnification) or 7 per mm2
- Myocarditis – the presence of an inflammatory infiltrate of a minimum of 14 infiltrating leukocytes/mm2
- Active myocarditis – the presence of > 14 infiltrating leukocytes/mm2 and/or the presence of more than 2 CD3+ T-lymphocytes per HPF, which are adherent to the contour of cardiomyocytes and focally associated with cell necrosis.
The most common cardiotropic viruses that cause myocarditis and DCMI (22):
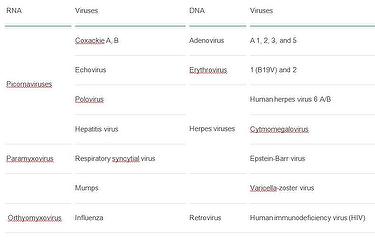
The current theory on the pathogenesis of DCMI states that the disease is a sequel of an inadequate (too weak or too strong) reaction of host immunologic system directed against microbial agent. More often DCMI is a result of a disproportionately strong reaction of the immunologic system, which escapes from the control mechanisms despite numerous “switching off” signals. In the prolonged low-level inflammation, cardiomyocytes are phagocytised, surrounded by various pathologic auto-antibodies, and pushed towards apoptosis. There is imbalance of pro-inflammatory cytokines, such as tumor necrosis factor (TNFα) or interleukins – IL-6 and IL-17 over anti-inflammatory cytokines tumor growth factor (TGFβ) or interferon (IFNγ) (23). As a result, the number of cardiomyocytes is greatly reduced.
III - Three-staged diagnostic process
To structure and simplify the DCM patient flow, a three-staged diagnostic algorithm has been proposed and is presented below.
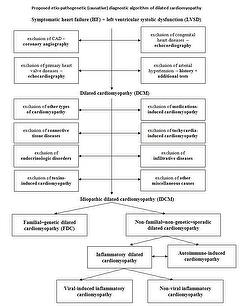
Stage I – Screening
- Exclusion of coronary artery disease (CAD)
Significant CAD is most commonly defined as > 50% stenosis of at least one principal coronary artery. - Exclusion of primary valvular heart disease or congenital heart disease
This should be performed by history taking, medical records, physical examinations and especially by transthoracic echocardiography and if necessary by - Exclusion of arterial hypertension
Exclusion of hypertension is probably the most challenging in this initial diagnostic process, especially in individuals more advanced in age. Tools to verify the possible role of hypertension in the pathogenesis of DCM should be a careful patient history, including the number of drugs and the dosage of antihypertensive medications, previous hospital records, as well as electrocardiographic and echocardiographic signs of LV hypertrophy.
Stage II – Identification of “diagnosable” causes
- Other forms of cardiomyopathies that may mimic and/or lead to DCM include: end-stage HCM, RCM, peripartum cardiomyopathy, stress-induced cardiomyopathy (Tako-tsubo syndrome), left ventricular non-compaction (24- 26). The presence of distinct features of the aforementioned types of cardiomyopathies is verified by comprehensive echocardiography and CMR.
- Connective tissue diseases include systemic lupus erythematosous, scleroderma, giant cell arthritis, etc. Exclusion process depends mostly on typical features of connective tissue disorders as well as the presence and titer of antinuclear and other disease-specific antibodies.
- Endocrinologic disorders that may rarely lead to the phenotype of DCM include thyroid hormone excess or deficiency, pheochromocytoma, Cushing’s disease.
- Toxins-induced cardiomyopathy – the most common toxin is ethanol following by illegal drug abuse of cocaine and amphetamines. Other rare toxins include cobalt, lead, mercury. Detailed patient’s history is the most important component.
- Medications-induced cardiomyopathy – the most pathogenic are chemotherapeutic agents such as anthracyclines, cyclophosphamide, and trastuzumab. Additionally, antiretroviral drugs (against HIV infection) such as zidovudine, didanosine, and zalcitabine are proven drugs implicated in DCM. Less evidence exists for other medications phenothiazines, chloroquine, and clozapine.
- Tachycardia-induced cardiomyopathy – persistent tachycardia, particularly uncontrolled atrial fibrillation, atrio-ventricular nodal reentry, and preexcitation syndromes with ventricular rates of 130 to 200 beats per minute may lead to DCM.
- Infiltrative disease – include amyloidosis, sarcoidosis, and hemochromatosis. Amyloidosis produces typical signs on ECG (low voltage in the limb and chest leads and conduction abnormalities), echocardiography (prominent diastolic dysfunction, symmetric LV wall thickening, and a granular, “sparkling” appearance of the myocardium), and amyloid deposits on myocardial bioptates. Diagnosis of sarcoidosis depends on the multi-system features of sarcoidosis and the evidence of non-caseating granulomas isolated either from the myocardium or other affected tissue.
- Hemochromatosis should be suspected by the presence of an elevated serum transferrin saturation and confirmed after typical findings of very low signal intensity on CMR and/or detected iron deposits on cardiac samples.
- Miscellaneous causes of cardiomyopathy include neuromuscular diseases (Duchenne’s muscular dystrophy, myotonic dystrophy, Friedreich’s ataxia), neoplastic heart disease (primary heart tumors, metastases to heart), celiac disease, extensive chest radiation, nutritional deficiencies (thiamine, selenium, and L-carnitine), obstructive sleep apnea (27). Those causes are very rare, nevertheless when suspected, appropriate diagnostic pathways should be followed.
At the end of Stage II, DCM will be classified as idiopathic dilated cardiomyopathy as no other detectable causes of DCM have been identified.
Stage III – Establishing the etiology of idiopathic dilated cardiomyopathy
Appropriate examinations such as echocardiographic studies of family members, genetic studies, and histopathological/immunochemistry/molecular biology studies of cardiac samples should be performed to differentiate between two main causes of idiopathic DCM such as genetic and inflammation/autoimmunisation.
Conclusions
A simplified three-staged diagnostic pathway has been proposed. After exclusion of the relatively rare causes of DCM, such as endocrinologic or autoimmune disorders, there are two wide groups of remaining conditions – genetic DCM and inflammatory DCM which are usually a distant sequel of myocarditis. In specialised cardiomyopathy centers, sophisticated in-depth diagnosis of genetic and inflammatory causes of DCM are slowly becoming a reality. Unfortunately, at present evidence-based and cost-effective causative treatments of genetic or inflammatory DCM remain difficult. This is however a very dynamic field and with possible therapeutic improvements.
The point is not to advocate complicated and expensive tests for every DCM patient, nevertheless, this is a very dynamic field and we should be aware of the growing diagnostic potential and possibly novel effective treatment that may be just around the corner for what were once “no hope” patients.
The content of this article reflects the personal opinion of the author/s and is not necessarily the official position of the European Society of Cardiology.